Download

1 / 46
460 likes | 865 Views
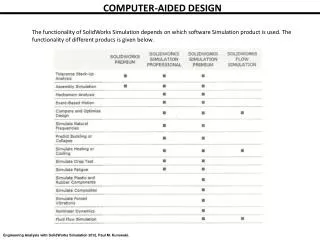
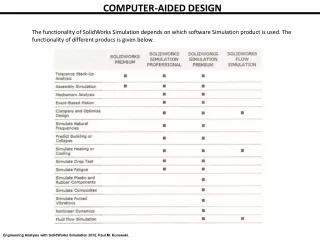
Computer-aided Color Chemistry. Methodology & Theory Practical issues. David A. Gallagher Portland, Oregon, USA David@CACheResearch.com. *. 2. 0. -2. -4. -6. MO Energy eV*. -8. . . -10. . Empty. Occupied. -12. 40000. 30000.

E N D
Computer-aided Color Chemistry • Methodology & Theory • Practical issues David A. Gallagher Portland, Oregon, USA David@CACheResearch.com *
2 0 -2 -4 -6 MO Energy eV* -8 -10 Empty Occupied -12 40000 30000 Molar Absorptivity PABA 20000 l/mol-cm 10000 0 200 220 240 260 280 300 320 340 360 380 400 Wavelength (nm) Mechanism of UV Absorption LUMO p-aminobenzoic acid (PABA) HOMO *quantum mechanics calculation (EHT, MOPAC,ZINDO, etc.)
-6 Transition instantly perturbs all orbitals -7 -8 -9 -10 ZINDO MO Energy eV -11 -12 -13 40000 Configuration Interaction (C.I.) -6 -7 -8 -9 Simple HOMO-LUMO gap over-estimates transition energy C.I. allows for energy contribution from relaxation of orbitals -10 Extended Huckel MO Energy eV -11 -12 -13 40000 *Configuration Interaction (C. I.) improves accuracy
ZINDO Parameters Spectroscopic Geometry INDO/1 parameters INDO/S parameters Zerner’s Intermediate Neglect of Differential Overlap (ZINDO) Semiempirical quantum chemistry method Ridley, Zerner , Theoret. Chim. Acta. 32, 111 (1973)
2 0 -2 -4 -6 MO Energy eV -8 -10 -12 40000 30000 Molar Absorptivity PABA 20000 l/mol-cm 10000 0 200 220 240 260 280 300 320 340 360 380 400 Wavelength (nm) Visualization of Chromophore *Location & structure aids design of dye
Absorption Intensity ~ Overlap Final orbital Initial orbital Absorption intensity depends upon the overlap of the initial and final orbitals (transition dipole).
Reality ZINDO approximation Vibrational Fine Structure
high gradient* ~ broad band 15 15 10 10 5 5 0 0 -5 -5 -10 -10 1.1 1.6 2.1 2.6 3.1 3.6 1.1 1.6 2.1 2.6 3.1 3.6 Higher gradient allows access to more vibration levels from ground state and hence, higher band width Gradient Affects Band Width low gradient* ~ narrow band
Band Width Estimation Higher gradient allows access to more vibration levels from ground state and hence, higher band width Gradient calculated from the “excited state energy” of the ground state geometry
Fluorescence Spectrum k < 109 sec-1 vibrational relaxation k > 1012 sec-1 S1 S0 S0 S1 excited singlet geometry Fluorescence Spectra UV-visible Absorpton Spectrum k = 1015 sec-1 ground state geometry
Calculated Absorption & Fluorescence Spectra Fluorescein (dibasic) Spectra UV-visible* λmax ~ 500nm Fluorescence* λ~ 535nm fluorescein2- *H. Du, R. A. Fuh, J. Li, A. Corkan, J. S. Lindsey, "PhotochemCAD: A computer-aided design and research tool in photochemistry," Photochemistry and Photobiology, 68, 141-142, 1998
Calculating Spectra with CAChe UV-visible Absorption Property of: “chemical sample” Property: “UV-visible transitions” Using: “ZINDO CI at PM3 geo...” Fluorescence Emission Property of: “excited states” Property: “optimized geometry” Using: “PM3/CI........geometry” Property of: “chemical sample” Property: “UV-visible transitions” Using: “current geometry”
Calibrate before use Safe Laboratory Procedure • Is the instrument working? • Do I know how to use it? • Is this instrument appropriate for the current research? Questions answered:
500 observed = - 41.157 + 1.0996 calculated R^2 = 0.998 400 . Observed absorption maximum (nm) 300 at INDO/1 geometry at MOPAC AM1 geometry R^2 = 0.998 Observed = - 45.277 + 1.1500 Calculated 200 200 300 400 500 Calculated absorption maximum (nm) Experimental vs Calculated UV/Vis Absorption Maxima for CH3-(CH=CH)n-CHO
400 n -> p* 380 360 340 320 200 220 240 260 280 300 n ->p* Transition of C=O CH3CHO acetaldehyde in hexane CH3COCH3 acetone in hexane CH3COCl acetyl chloride in hexane CH3COOCH2CH3 ethyl acetate in water CH3CONH2 R^2 = 0.988 acetamide in water ZINDO = 188.00 + 0.69698 obs CH3COOH acetic acid Standard Deviation = 2.8 nm in ethanol Observed *error bars represent bandwidth
Anthraquinones for Thermal Transfer Wax Printing Dyes used in thermal wax printing often behave differently than they do on fabrics. Can computer-aided chemistry provide useful information about dyes that might be used in wax inks? Intratherm Blue P1404 Dr. Wayne Jeager, Tektronix, Inc., Wilsonville, Oregon, USA.
Molar Absorptivity 25000 20000 Molar Absorptivity (l/mol-cm) 1,4-amino anthraquinone 15000 25000 10000 20000 5000 1,4-amino anthraquinone 15000 1,4-diNHMe aq Exp 0 10000 350 400 450 500 550 600 650 700 750 5000 Wavelength (nm) 0 350 400 450 500 550 600 650 700 750 Wavelength (nm) • First attempt • Experimental Why doesn’t the calculated spectrum agree with the measured spectrum? Calculated vs. Experimental *
exp max 700 10 4 600 11 7 8 500 9 2 6 1 400 5 3 300 430 440 450 460 470 480 490 calculated max exp -max =3.834 *calculated -max -1251.676 r ^ 2 = 0.430 Anthraquinone Calibration 1 anthraquinone 2 1-NH2 anthraquinone 3 1-OH anthraquinone 4 1,4-diNH2 anthraquinone 5 2-NH2 anthraquinone 6 2-NMe2 anthraquinone 7 1-NHEtNMe2 anthraquinone 8 1,9-HNEtNMe2 anthraquinone 9 1,6-HNEtMe2 anthraquinone 10 1,4-diNHMe anthraquinone 11 1,4-diNH2, 2-OMe aq Experimental data from Masafumi Adachi and Shinichiro Nakamura, Dyes and Pigments, 1991, 17, 287-296, and Wayne Jaeger, Tektronix.
What’s Wrong? • Is ZINDO right? • What is ZINDO calculating? • Are we comparing the right absorptions? • Is solvation a factor? • Is aggregation a problem? • Are the structures right? • Which is best geometry/conformation to use? • Experimental data correct?
Is ZINDO right? • literature • absolute accuracy • relative accuracy • zero-point energy corrections
Literature Background • “The literature suggests that at visible wavelengths the standard deviation of the [INDO/S] calculation is between 15 and 35 nm. • “-maxima calculations using INDO seem useful when the geometries including dihedral angles are correct.” Louis E. Friedrich and James E. Eilers, “Progress Toward Calculation of the Hues of Azomethine Dyes”, J. of Imaging Science and Technology, 38, 24-27, 1994. [Eastman Kodak Company, Rochester, NY]
600 4-NEt , 2',4',6'-triCN 2 4-NEt , 4'-NO 500 2 2 N N 2-NH 2 400 Exp_max = - 534.52 + 2.7595 Calc_max R^2 = 0.912 300 300 320 340 360 380 400 ZINDO max R 600 R' Exp_max = 79.664 + 0.84424 Calc_max R^2 = 0.947 C p-NO C H ; -CH ;Anion 2 6 4 3 N H -H;-H;Anion 500 N p-NH C H ; -CH ;Anion 2 6 4 3 NO -H;-H;Anion 2 p-NH C H ; -CH ;Neutral 2 6 4 3 400 . p-NO C H ; -CH ;Neutral 2 6 4 3 -H;-H;Neutral 300 NO 300 400 500 600 2 ZINDO max Dye Calibrations Azobenzene Dyes Hydrazone Dyes Data from Masafumi Adachi and Shinichiro Nakamura, Dyes and Pigments, 1991, 17, 287-296. Experiment based geometries were used.
NMe 2 NMe 2 O + 6 2 O 4 600 700 2,3-diCN,4-NH Obs_max = - 569.41 + 2.5311Calc_max 2 R^2 = 0.928 2,3-diCl;5-NH ;-8-OMe 2 4-CN . . 3-CN None 500 5-NH ,8-OMe 600 max 2 max 5-NH l 2 l Exp Exp 400 500 . . 5-OMe None 3,5-diNO Obs_max = - 329.17 + 1.9616 Calc_max 2 R^2 = 0.881 4-CN 300 400 360 380 400 420 440 460 380 400 420 440 460 480 500 520 l ZINDO l ZINDO max max More Dye Calibrations Napthoquinone Dyes Cationic Dyes Data from Masafumi Adachi and Shinichiro Nakamura, Dyes and Pigments, 1991, 17, 287-296,
1 benzene 2 naphthalene 3 anthracene 4 naphthacene 5 pentacene Linear Fused Aromatic Rings 700 600 5 500 exp lambda max 4 400 3 300 2 200 1 100 0 1 2 3 4 5 6 # aromatic rings experimental lambda max value =99.200 *# aromatic rings+ 74.600 r ^ 2 = 0.997
What Geometry Did We Calculate? Lowest energy structure from MOPAC/PM3 Heat of formation = -15.35 kcal/mol Dihedral O-C-C-C = 158° Expected structure Heat of formation = -11.27 kcal/mol Dihedral O-C-C-C = 180°
Does the Geometry Matter? • First attempt • Flattened rings
Which structure is right? Crystal structure of N,N’-diphenylaminoanthraquinone (solid) 6-31G* LDF structure of 1,4-diaminoanthraquinone (gas-phase)
Indigo model lambda max visible (nm) PM3 413.2 AM1 400.4 6-31G*LDF 453.3 observed 546.0 6-31G* LDF using BLYP and Mulliken. Observed value from P. W. Sadler, March, 1956. 50000 40000 30000 indigo@6-31G*LDF ZCI 20000 10000 0 250 300 350 400 450 500 550 600 650 Wavelength (nm) Sensitivity to Model Indigo 6-31G* LDF (darker) vs AM1 (faded)
CPU time Indigo model PM3 00:52 00:35 AM1 1d 11:05:00 6-31G*LDF PM3 and AM1 using MOPAC 6-31G* LDF using BLYP and Mulliken. Processing Times Indigo
Force a flat structure Lock dihedral angles to force a flat structure
Molar Absorptivity (l/mol-cm) 25000 20000 15000 1;4-diNHMe anthraquinone 1;4 amino anthraquinone 10000 1;4-diNHMe aqF Exp 5000 0 350 400 450 500 550 600 650 700 750 Wavelength (nm) Effect of Ring Geometry • First attempt • Flattened rings • Experimental
Molar Absorptivity (l/mol-cm) 25000 20000 1;4 amino anthraquinone 1;4-diNHMe anthraquinone 15000 1;4-diNHMe aqF 10000 1;4-diNHMe aqF Exp 5000 0 350 400 450 500 550 600 650 700 750 Wavelength (nm) Effect of Ring & Amine Geometry • First attempt • Flattened rings, • Flattened rings & amines • Experimental
Molar Absorptivity (l/mol-cm) 25000 20000 1;4-diNHMe aqF Exp 15000 1;4-diNHMe aqF 10000 1;4-diNHMe aqF/(MeOH)2 5000 0 350 400 450 500 550 600 650 700 750 Wavelength (nm) Methanol Solvation? • Gas-phase • Solvated • Experimental
exp max 700 1 anthraquinone 2 1-NH2 anthraquinone 6 600 3 1;4-diOH aq 7 4 4 1;4-diNH2 anthraquinone 5 500 3 5 1-NH2;2-OPh;4-OH aq 2 6 1;4-diNHMe aq 400 7 1;4-diNH2;2-OMe aq 1 300 200 300 400 500 calculated max Anthraquinone Calibration exp max =1.606 *calculated max -144.684 r ^ 2 = 0.958 cross validated against 5 samples with unsigned average error 20 nm Experimental data from Masafumi Adachi and Shinichiro Nakamura, Dyes and Pigments, 1991, 17, 287-296, and Wayne Jaeger, Tektronix.
Effect of Calibration Calibrated INDO/S at the PM3 geometry with a flat anthraquinone ring and flat amino groups. • First attempt • Flattened rings, • Flattened rings & amines • Flattened rings & amines, calibrated • Experimental
Conclusions • INDO/S can be calibrated so that it is useful for predicting UV/visible spectra of dyes. (ZINDO predictions are systematically too short.) • Calculated spectra are sensitive to molecular geometry. Best results are obtained with experimental geometries. • Vibrational structure can confuse spectral assignments. • Excited state gradients correlate with band widths.
Chemistry Today Properties Measure Design Structure Compounds Synthesize
Chemistry by Simulation Properties Simulate Design Structure Compounds Build
Chemistry by Design Properties Interpret Simulate Design Structure Compounds Build
Summary Points • You learn something almost every time you view a molecule a new way • Safe laboratory procedure: calibrate before use • Bootstrap from small problems to large problems • Stretch beyond simulation of experiment to interpretation. Valuable insight will come from understanding why experiments and calculations do not agree
ZINDO Capabilities • Contains d-orbitals. • Geometry parameters for first two transition metal series. • Spectroscopic parameters for UV/visible calculations through first transition metal series. • Can predict singlet-triplet splittings. • Contains self-consistent reaction field (SCRF) for inclusion of solvent effects on geometries.
ZINDO Limitations • Geometry parameters based on ab initio HF. • Zero differential overlap seriously over estimates the stability of small rings. • Optimization is too aggressive for transition metals, leading to the formation of small ring structures. • Transition metal INDO/1 parameters used for geometry optimizations are not sufficiently tested. • Absorption maxima for many dyes are systematically underestimated and calibration for each class of dyes is recommended. • Mono CI does not predict double excitation states. • SCRF is not used for spectral calculations.
Background References for ZINDO • ZINDO is a semi-empirical SCF/CI package including analytical gradient optimization developed by Prof. M. C. Zerner from the University of Florida. ZINDO is based upon the INDO approximations of Pople, Santry and Segal: • Pople, Santry, Segal J. Chem. Phys. 43, S129 (1965) • Pople, Segal J. Chem. Phys. 43, S136 (1965) • Pople, Segal J. Chem. Phys. 44, 3289 (1966) • Santry, Segal J. Chem. Phys. 47, 158 (1967) • Santry J. Amer. Chem. Soc. 90, 13 (1968) • The original INDO method has been enhanced by Prof. Zerner to include spectroscopic parameterization, configuration interaction, higher angular momentum orbitals (d-orbitals), and analytical gradients: • Ridley, Zerner , Theoret. Chim. Acta. 32, 111 (1973) • Ridley, Zerner, Theoret. Chim. Acta. 42, 223 (1976) • Ridley, Zerner, Theoret. Chim. Acta. 53, 21 (1979) • Zerner, Loew, Kirchner, Mueller-Westerhoff, J. Amer. Chem. Soc. 102, 589 (1980) • Head, Zerner, Chem. Phys. Lett. 32, 246 (1985) • Head, Zerner, Chem. Phys. Lett. 131, 359 (1986) • Anderson, Edwards, Zerner, Inorg. Chem. 25, 2728 (1986) • Edwards, Zerner, Theoret. Chim. Acta. 72, 347 (1987)
Applications of ZINDO • 1. J. Ridley and M. C. Zerner, Theoret. Chim. Acta (Berl.) 72, 111-134 (1973)"An Intermediate Neglect of Differential Overlap Technique for Spectroscopy: Pyrrole and the Azines". • 2. W. P. Anderson, W. Daniel Edwards, and Michael C. Zerner, Inorg. Chem. 1986, 25 2728-2732, "Calculated Spectra of Hydrated Ions of the First Transition-Metal Series." • 3. W. Daniel Edwards, Brian Weiner and Michael C. Zerner, J. Am. Chem. Soc, 1986, 108, 2196-2204, "On the Low-Lying States and Electronic Spectroscopy of Iron(II) Porphine." • 4. Ralph S. Becker, L. V. Natarajan, Christian Lenoble, and Ronald G. Harvey, J. Am. Chem. Soc. 1988, 110, 7163-7167, "Photophysics, Photochemistry and Theoretical Calculations of Some Benz[a]anthracene-3,4,-diones and Their Significance." • 5. Jikang Feng, Jerzy Leszczynski, Brian Weiner, and Michael C. Zerner, J. Am. Chem. Soc, 1989, 111, 4648-4655, "The Reaction C3H3+ + C2H2 and the Structural Isomers of C5H5+." • 6. Frank U. Axe, Charles Flowers, Gilda H. Loew, and Ahmad Walch, J. Am. Chem. Soc., 1989, 111, 7333-7339, "Theoretical Studies of High-, Intermediate-, and Low-Spin Model Heme Complexes."
Applications of ZINDO (cont) • 7. Manfred Kotzian, Notker Rosch, Hartmut Schroder, and Michael C. Zerner, J. Am. Chem. Soc., 1989, 111, 7687-7696, "Optical Spectra of Transition-Metal Carbonyls" Cr(CO)6, Fe(CO)5, and Ni(CO)4." • 8. Wayne P. Anderson, Thomas R. Cundari, Russell S. Drago, Michael C. Zerner, Inorg. Chem. 29, 1, 1990, "Utility of the Semiempirical INDO/1 Method for the Calculation of the Geometries of Second-Row Transition Metal Species." • 9. David R. Kanis, Mark A. Ratner, and Tobin J. Marks, J. Am. Chem. Soc., 1990, 112, 8203-8204, "Description of Quadratic Optical Nonlinearities for Transition-Metal Organometallic Chromophores Using an SCF-LCAO MECI Formalism." • 10. Mati Karelson and M. C. Zerner, J. Am. Chem. Soc., 1990, 112, 9405-9408, "On the n-š* Blue Shift Accompanying Solvation." • 11. Yuji Kubo, Katsuhira Yoshida, Masafumi Adachi, Shimichiro Nakamura, and Shuichi Maeda, J. Am. Chem. Soc. 1991, 113, 2868-2873, "Experimental and Theoretical Study of Near-Infrared Absorbing Naphthoquinone Methide Dyes with Nonplanar Geometry." • 12. Masafumi Adachi and Shinichiro Nakamura, Dyes and Pigments, 1991, 17, 287-296, “Comparison of the INDO/S and the CNDO/S Method for the Absorption Wavelength Calculation of Organic Dyes.”
Acknowledgements Thank you to George D. Purvis for providing some of the content for these slides .