Download
1 / 20
200 likes | 395 Views
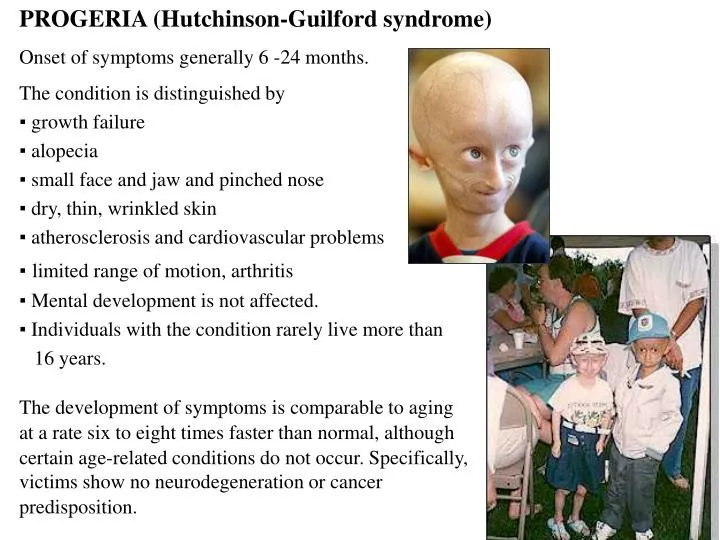
PROGERIA (Hutchinson-Guilford syndrome) Onset of symptoms generally 6 -24 months. The condition is distinguished by ▪ growth failure ▪ alopecia ▪ small face and jaw and pinched nose ▪ dry, thin, wrinkled skin ▪ atherosclerosis and cardiovascular problems

E N D
PROGERIA (Hutchinson-Guilford syndrome) Onset of symptoms generally 6 -24 months. The condition is distinguished by ▪ growth failure ▪ alopecia ▪ small face and jaw and pinched nose ▪ dry, thin, wrinkled skin ▪ atherosclerosis and cardiovascular problems ▪limited range of motion, arthritis ▪ Mental development is not affected. ▪ Individuals with the condition rarely live more than 16 years. The development of symptoms is comparable to aging at a rate six to eight times faster than normal, although certain age-related conditions do not occur. Specifically, victims show no neurodegeneration or cancer predisposition.
Progeria, the classic "accelerated aging disease" is not caused by defective DNA repair. It is caused by mutations in a LMNA (Lamin A protein) gene. Lamin A is part of a protein scaffold around the edge of the nucleus that helps organize nuclear processes such as DNA and RNA synthesis. Prelamin A is farnesylated, enabling it to bind to the nuclear membrane. The cleavage site to release Lamin A is mutated. Lamin A cannot be produced and Prelamin A builds up on the nuclear membrane
Bloom's Syndrome -typical physical characteristics:· Short stature · A narrow face with prominent nose · Skin color changes in the face. Change more noticeable after sunlight exposure · Butterfly-shaped facial rash, similar to rash caused by Lupus Erythematosis · A high pitched voice · An increased susceptibility to infections and respiratory illness · An increased susceptibility to cancer and leukemia · Some may also have mental retardation
Classical Cockayne syndrome (Type I) ∙ progressive symptoms become apparent after 1 year. ∙ unlike other DNA repair diseases, Cockayne syndrome is not linked to cancer
Xeroderma Pigmentosum (XP) condition resulting from a deletion in the gene for the XPAC protein of the excision repair system, so that individuals are unable to reverse T^T dimerization induced by sunlight. XP is characterized by extremely dry skin and numerous malignant pinpoint tumors induced after brief exposure to sunlight. The boy on the left shows two skin cancers on his nose, and an irregular distribution of pigment that resembles a severe sunburn (erythemea). The girl on the right has avoided sunlight since diagnosis with XP.
UV-protection suit to help people suffering from the genetic disorder xeroderma pigmentosum (XP). It consists of a headgear protecting the head and face, and a suit covering the rest of the body, both filtering out 100% UV-light from the Sun
DYSKERATOSIS CONGENITA Proposed model of the pathogenesis of bone marrow failure in patients with DKC In this model excessively short telomeres are the underlying cause of clinical manifestations in patients with DKC. Mutations in TERC or TERT affect the activity of telomerase. Mutations in DKC1 destabilize TERC and/or retard ribosome biogenesis. As telomeres get shorter there is an increase in genomic instability leading eventually to a cell crisis, cell cycle arrest, and cell death. Rare cells that can maintain telomere integrity emerge from the crisis as potentially malignant cells. From: Mason PJ, Wilson DB, Bessler M Dyskeratosis congenita -- a disease of dysfunctional telomere maintenance. Curr Mol Med 2005 Mar;5(2):159-70
Leukoplakia-- white plaques or patches on the mucous membranes of the oral cavity, including the tongue • Dyskeratosis congenita • Clinical features: • ● abnormal skin pigmentation • ● leukoplakia • ● nail dystrophy • ● bone marrow failure Reticulated pigmentation seen in 90% of patients
Dyskeratosis congenita: Disintegrating nails appear between ages 5-13 years and are eventually destroyed.
● 75% mortality by age 30. ● Bone marrow failure is a major cause of death, with approximately 75% of deaths related to bleeding and opportunistic infections as a result of peripheral cytopenia.




















