Download

1 / 19
190 likes | 350 Views
Structural Classification and Prediction of Reentrant Regions in Alpha-Helical Transmembrane Proteins: Application to Complete Genomes Håkan Viklunda, Erik Gransetha and Arne Elofsson Journal of Molecular Biology 2006 Aug 18;361(3);591-603. Tim Nugent BugF 8 th March 2007.

E N D
Structural Classification and Prediction of Reentrant Regions in Alpha-Helical Transmembrane Proteins: Application to Complete Genomes Håkan Viklunda, Erik Gransetha and Arne Elofsson Journal of Molecular Biology 2006 Aug 18;361(3);591-603. Tim Nugent BugF 8th March 2007
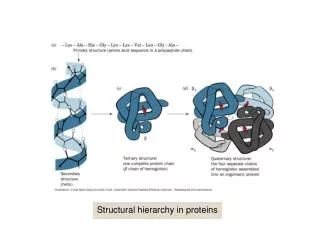
Structural regions of alpha-helical proteins • Recently, the number of solved alpha-helical TM structures has increased rapidly. • Structural complexity has been revealed to be equivalent of globular proteins. • The most prominent features of TM proteins are membrane spanning alpha-helices. • These are connected by loop regions.
Substructures • Several other functionally and structurally important substructures exists. • One such substructure is the interface helix region, situated parallel with the membrane in the membrane-water interface region. • Another type is the reentrant region – part of the loop region which penetrates the membrane, but enters and exits on the same side.
Definition and properties or reentrant regions • Reentrant regions are defined as sequences which start and end on the same side of the membrane, and penetrate between 3 Å and 25 Å. • Sequence stretches with a depth of between 1.5 Å and 3 Å are also defined as reentrant regions if residue depth monotonically increase/decrease on the respective entrance/exit sides of the deepest residue, and there is a clear turn in the membrane. • Classification was performed by visual inspection. • 79 transmembrane proteins with known 3D structure were attained from the Membrane Protein Structure database and the Protein Data Bank. Homology reduced at 30% sequence similarity. • Based on the definition: • 36 reentrant regions • 302 transmembrane regions • 80 interface helix regions
Region comparison • Fraction of irregular secondary structure elements is larger in reentrant regions than in regular TM helices. • Average fraction of helical residues for reentrant regions is 57% with a clear correlation between helical content and length of the region (correlation coefficient = 0.75).
Three classes of reentrant regions can be identified • Based on secondary structure - a helix must be at least 5 residues long; shorter helical regions are defined as a coil. • Helix-Coil-Helix:
Three classes of reentrant regions can be identified • Helix-Coil or Coil-Helix:
Three classes of reentrant regions can be identified • Coil / irregular secondary structure:
Identification and prediction of reentrant regions • Developed TOP-MOD - a hidden Markov model-based method to classify the residues of a TM sequence into four structural classes – M, R, I and L.
Distinguishing reentrant regions from loop and interface helix regions • Believed that reentrant regions form relatively late in the overall folding dynamics, after the initial translocation and formation of the membrane spanning helices. • Their emergence can be visualised as a process in which parts of inter-TM regions are pulled into the membrane. • To test this, inter-TM parts from each sequence were cut out and TOP-MOD was used to make a region classification on these subsequences.
Distinguishing reentrant regions from loop and interface helix regions
Predicting reentrant regions on whole sequence level • So far, TOP-MOD has only been tested on sequences connecting TM helices. • The possibility to distinguish between different types of structural region on a whole sequence level was evaluated. • First, sequences where the approximate location of TM regions was considered to be know were analysed. Central residues of membrane regions were constrained to the HMM compartment modeling the membrane regions using sequence labels. • Second, topology predictor PRODIV-TMHMM used as a pre-processor to predict location of TM helices.
Scanning for reentrant regions in E. coli, S. cerevisiae and H. sapiens • Using TOP-MOD and PRODIV-TMHMM, TM proteins of E. coli, S. cerevisiae and H. sapiens were scanned to make a preliminary estimate of the occurrence of reentrant regions in these genomes. • Fraction is found to be at least 10% in all three genomes. • To avoid false positives, sensitivity was set fairly low suggesting that the reentrant fraction may be even higher.
Scanning for reentrant regions in E. coli, S. cerevisiae and H. sapiens • Fraction of proteins predicted with reentrant regions increases linearly with the number of predicted TM regions. • In two TM-number categories the fraction is lower: 7-TM GPCRs and 12-TM major facilitator superfamily transporters.
Proteins of a particular molecular function with predicted reentrant region • Each sequence was mapped to HMM-based domain library PFAM. • Earlier literature suggests reentrant loops were primarily found in passive transporter proteins. • This data suggests their occurrence in active transporters is higher than previously thought.
Conclusions • For at least the last 10 years, the dominating non-experimental way of attaining structural information of alpha-helical TM proteins has been by predicting topology. • As more 3D structures have been resolved, it has become apparent that TM proteins are often too complex to fit in to the helix, inside loop, outside loop constraints where loops are always on opposite sides of the membrane. • This suggests that a finer grained nomenclature, as well as finer grained methods, is needed to study these proteins. • Define more detailed substructures. • Predict the structure directly using ab initio methods. • Solve more 3D structures.