Download

1 / 103
1.07k likes | 1.33k Views
METABOLISM of AMINO ACIDS Dr Shahnaz Khaghani. Tehran University of Medical Sciences. TRANSAMINATION (Amino nitrogen ). CATABOLISM pyruvate ace tyl-CoA acetoacetate succinyl-CoA, α -KG. GLUTAMATE. AMINO ACIDS. UREA. Anabolic pathways Specialized molecules: creatine,

E N D
METABOLISM ofAMINO ACIDSDr Shahnaz Khaghani Tehran University of Medical Sciences
TRANSAMINATION (Amino nitrogen ) CATABOLISMpyruvateacetyl-CoAacetoacetatesuccinyl-CoA, α-KG GLUTAMATE AMINO ACIDS UREA • Anabolic pathways • Specialized molecules: • creatine, • SAM, cysteine, hormones,NO, • dTMP ABERRANT METABOLISM PKU, MSUD
OUTLINE • Incorporation of amino acid nitrogen into urea • A) Transamination • B) Glutamate dehydrogenase generates ammonia • C) Urea synthesis requires ammonia, • bicarbonate, and ATP • 1) hyperammonemia, cause and management • Catabolism (utilization) of amino acid carbon atoms • A) Amino acid carbon atoms pyruvate, thus gluconeogenesis
III. Amino acids as substrates in critical anabolic pathways A) Creatine (arginine and glycine) B) Hormones: catecholamines (phenyl- alanine), serotonin (tryptophan), thyroid hormones (phenylalanine) C) “Second messenger,” nitric oxide (NO) (arginine) D) Methyl donor, S-adenosyl-L- methionine or SAM(methionine) E) Cysteine (methionine and serine) F) Methionine resynthesis and DNA synthesis
IV. “Inborn errors” of amino acid metabolism A. Phenylketonuria 1) Clinical Manifestations: a) growth failure b) delayed psychomotor development c) seizures d) mental retardation
Examples, “inborn errors” of amino acid metabolism • A. PKU • B. MSUD
B) Amino acid carbon atoms acetyl-CoA, thus fatty acids and cholesterol C) Amino acid carbon atoms acetoacetate, thus “ketone bodies” D) Amino acid carbon atoms succinyl-CoA, thus TCA cycle E) Amino acid carbon atoms 2-oxoglutarate (α-KG), thus TCA cycle
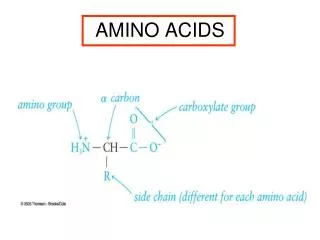
Amino Acid Breakdown: No storage form of Amino Acids, therefore excess need to be converted to other forms to be used as energy or stored as glycogen/fat. R O H2N CH C OH Recycling of the Carbon Skeleton Disposal of Nitrogen Atom (Urea)
I. Amino Acid Nitrogen Incorporation into Urea A. TransaminationTransamination requires: 1) An amino acid and an oxo- (keto-) acid 2) Co-factor pyridoxal phosphate 3) An aminotransferase. NH2 O O O | | | | | | | R1CH– COH+ R2 C– C– OH O O NH2 O | | | || | | R1– C – C– O H + R2–CH – C – OH
Flow of Nitrogen: In tissues (e.g Muscle), most amino acids transfer their a-amino group to Glutamate Aminotransferase B6 a-ketoglutarate
Biosynthesis of Amino Acids: Transaminations Amino Acid1 +a-Keto Acid2 Amino Acid2 +a-Keto Acid1 + Glutamate Pyridoxal phosphate (PLP)- Dependent Aminotransferase + a-Ketoglutarate
Transaminations: Role of PLP Tautomerization
The N is then transferred from Glutamate to Pyruvate, producing Alanine. a-ketoglutarate Pyruvate Aminotransferase
Four of the amino acids, Glycine, Lysine, Threonine and Serine are directly deaminated. Serine Dehydratase
NH3 released from Glycine/Lysine/Threonine/Serine is incorporated into Glutamine [ ] Glutamine Synthetase
In sum: During amino acid breakdown, the a -amino Nitrogen gets incorporated as the a-amino group in Alanine or the amide group in Glutamine. Alanine and Glutamine are then released to the circulation.
Flow of Nitrogen: Alanine and Glutamine released by peripheral tissues are taken up by the Liver. The Nitrogen on Alanine is transferred to a-ketoglutarate to produce Glutamate
a-ketoglutarate Glutamate Aminotransferase
Glutamate has two fates important for disposal of waste N. Conversion to a-ketoglutarate by Glutamate Dehydrogenase to release NH3 2) As N donor in the transamination of oxaloacetate to Aspartate
Conversion to a-ketoglutarate by Glutamate Dehydrogenase to release NH3 [ ] Glutamate dehydrogenase
2) As N donor in the transamination of oxaloacetate to Aspartate Glutamate -ketoglutarate O NH2 O O O O B6 HO-C-CH2-CH-C-OH HO-C-CH2-C-C-OH Aminotransferase Oxaloacetate Aspartate
Glutamine is hydrolyzed by Glutaminase to release NH3 Glutaminase NH3
Nitrogen flow in Liver Alanine Glutamate Aspartate NH3 Glutamine NH3
The oxoacid can be either pyruvate, which produces alanine, oxaloacetate, which produces aspartate 2-oxoglutarate, which producesglutamate
Transamination with 2-oxoglutarate yields glutamate. Glutamate yields ammonia. Ammonia enters the urea cycle. NH2 O | | | R– CH– C– O H + 2-OG O NH2 O | | | | | HO– C – CH2 – CH2 –CH – C– OH NH3
B. Glutamate dehydrogenase Glutamatetoammonia catalyzed by glutamate dehydrogenase (uses NAD+) O NH2 O | | | | | HO–C– CH2– CH2– CH - C – OH 2-oxoglutarate + NH3
C. Urea cycle Rate-determining step of the urea cycle requires ammonia, catalyzed by carbamoyl phosphate synthetase. NH3 + HCO3- + 2 ATP CARBAMOYL PHOSPHATE+ Pi + 2ADP
There are two different enzymes with this name. The urea cycle enzyme is carbamoyl phosphate synthetase I or CPS I
Carbamoyl phosphate contains nitrogen from ammonia (amino acids), carbon from bicarbonate, and phosphate from ATP. O O │ │ │ │ 2 H N - C - O - P - OH │ OH
Next, carbamoyl phosphate reacts with ornithine in a reaction catalyzed by ornithine carbamoyl transferase. Carbamoyl phosphate + ornithine citrulline
Citrulline reacts with aspartate to yield argininosuccinate. Second nitrogen of urea from aspartate. citrulline + aspartate argininosuccinate synthetase argininosuccinate + AMP + PPi
Argininosuccinate is cleaved by arginino-succinate lyase to yield arginine and fumarate. argininosuccinate arginine + fumarate
Final reaction, arginine is cleaved by arginase to yield urea and regenerate ornithine (it’s a cycle!) arginine urea+ ornithine
CARBAMOYL PHOSPHATE + UREA Ornithine Citrulline Arginine + UREA CYCLE Aspartate Fumarate Argininosuccinate
REGULATION OF THE UREA CYCLE Acute: N-acetylglutamate, allosteric effector, up regulates CPS I
N-Acetylglutamate is synthesized from glutamate and acetyl-CoA by a mitochondrial NAG synthase.
Metabolic Diseases of the Urea Cycle Arginase Deficiency Argininosuccinic acidemia Type II Hyperammonemia: Type I Citrullinuria
Metabolic Diseases of the Urea Cycle Disorders present in infants: Symptoms: Lethargy, swelling of the brain leads to mental retardation/brain damage Diagnosis: Low blood urea nitrogen (BUN) levels -high levels of ammonia in the blood elevated circulating glutamine -other metabolites that accumulate depend on the specific enzyme defect Most common form: Hyperammonemia Type II caused by Ornithine Transcarbamylase deficiency Elevated Carb-P levels in this deficiency cause secondary problems in pyrimidine metabolism
Treatment: Long term, dietary restriction. Low protein diet. Supplemented with Arginine Short term Dialysis Administration of Nitrogen “scavengers” e.g. Phenylacetate
Excessive ammonia is toxic to the central nervous system. Alternative pathway therapy: Sodium benzoate to produce hippuric acid Sodium phenylacetate or phenyl-butyrate to produce phenylacetylglutamine
NH3 + CO2 + 5,10-methylenetetra- hydrofolate NH2O|| |Glycine CH2 – C – OH Sodium Benzoate Bz-N-H O | | | CH2 – C - OH Benzoyl glycine or hippuric acid excreted
NH3 + glutamate + ATP → glutamine + ADP + Pi sodium phenylacetate + CoA → phenylacetyl-CoA Then glutamine + phenylacetyl-CoA → phenylacetylglutamine (excreted)
sodium phenylbutyrate + CoA → phenylbutyryl-CoA phenylbutyryl-CoA undergoes β-oxidation → phenylacetyl-CoA As above, phenylacetyl-CoA + glutamine→ phenylacetylglutamine (excreted)
II. Catabolic Fates of Amino-acid Carbon Atoms Post-transamination carbon atoms become those of pyruvateacetoacetate acetyl-CoA succinyl-CoA 2-oxoglutarate
Phenylalanine Methionine (partial) Leucine Tyrosine (50% of carbons) Cysteine Glycine Alanine Serine Aspartate Asparagine Tyrosine (50% of carbons) Phenylalanine Isoleucine (partial) Valine (partial) Methionine(partial) Glutamate Glutamine Proline Acetyl CoA GLUCOSE Pyruvate Oxaloacetate Citrate Malate Isocitrate Fumarate Succinate -Ketoglutarate Succinyl CoA Amino acids discussed in previous lectures Amino acidsto be discussed in this lecture Carbon end products from the degradation (catabolism) of amino acids