Download
1 / 44
440 likes | 698 Views
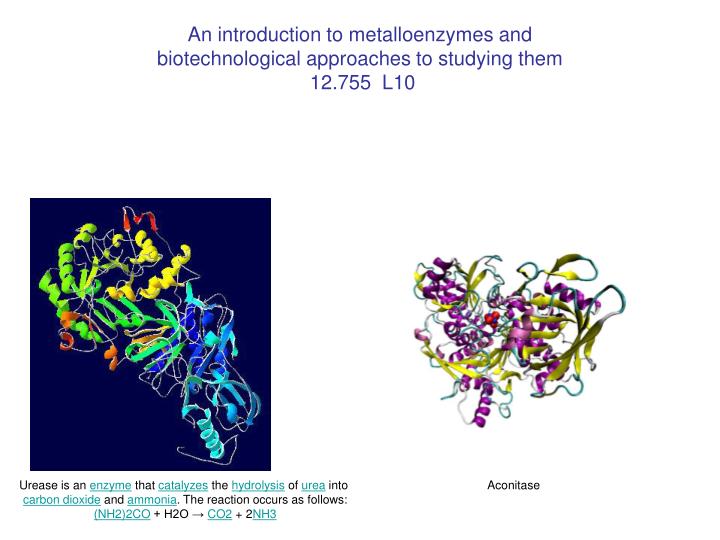
An introduction to metalloenzymes and biotechnological approaches to studying them 12.755 L10. Urease is an enzyme that catalyzes the hydrolysis of urea into carbon dioxide and ammonia . The reaction occurs as follows: (NH2)2CO + H2O → CO2 + 2 NH3. Aconitase. Outline.

E N D
An introduction to metalloenzymes andbiotechnological approaches to studying them 12.755 L10 Urease is an enzyme that catalyzes the hydrolysis of urea into carbon dioxide and ammonia. The reaction occurs as follows: (NH2)2CO + H2O → CO2 + 2NH3 Aconitase
Outline • Introduction – global BGC to cellular physiology to metalloenzyme and molecular • Categories of metalloprotein and metalloenzymes functions • The code: amino acids • The Genomic Firehose • Bioinformatic terminology • Intergrated Microbial Genomics Portal
Roles of metal in biology (From Bioinorganic Chemistry, Lippard and Berg) Metalloprotein Functions • Dioxygen Transport • Hemoglobin-myoglobin family • Hemocyanins • Hemerthyrins • Electron Transfer (e.g. nitrogen fixation) • Structural Roles (zinc fingers) Metalloenzyme Functions (Note: Metalloenzymes are metalloproteins that perform a catalytic function) • Hydrolytic Enzymes (Carbonic Anhydrases) • Two Electron Redox Enzymes (Nitrate Reductase, oxidation of hydrocarbons by P-450) • Multielectron Pair Redox Enzymes (Cytochrome c, PSII, Nitrogenase) • Rearrangements (Vitamin B12)
Key enzyme in the nitrification reaction:ammonia (NH3) hydroxylamine (NH2OH) nitrite (NO2-)Found in anaerobic oxidizing bacteria (AOB) but not the more abundant anaerobic oxidizing archaea (AOA)24 hemes (irons) per molecule!What does nature actually use in the oceans if this enzyme is not present?
How does a particular amino acid sequence create the function of a metalloprotein or the activity of a metalloenzyme?
“The sequence itself is not informative; it must be analyzed by comparative methods against existing databases to develop hypothesis concerning relatives and function.“ Terminology for comparing sequences: • Identity: The extent to which two (nucleotide or amino acid) sequences are invariant. • Similarity: The extent to which nucleotide or protein sequences are related. The extent of similarity between two sequences can be based on percent sequence identity and/or conservation. In BLAST similarity refers to a positive matrix score. • Conservation: Changes at a specific position of an amino acid or (less commonly, DNA) sequence that preserve the physico-chemical properties of the original residue. • Homology - Similarity attributed to descent from a common ancestor. NOTE: it is binary, sequences have homology or they do not. Something cannot be “highly homologous” • Source: http://www.ncbi.nlm.nih.gov/Education/BLASTinfo/glossary2.html
BLAST Basic Local Alignment Search Tool • E-Values: Expectation value. The number of different alignments with scores equivalent to or better than S that are expected to occur in a database search by chance. The lower the E value, the more significant the score. • In the limit of sufficiently large sequence lengths m and n, the statistics of HSP scores are characterized by two parameters, K and lambda. Most simply, the expected number of HSPs with score at least S is given by the formula The parameters K and lambda can be thought of simply as natural scales for the search space size and the scoring system respectively. • We call this the E-value for the score S. This formula makes eminently intuitive sense. Doubling the length of either sequence should double the number of HSPs attaining a given score. Also, for an HSP to attain the score 2x it must attain the score x twice in a row, so one expects E to decrease exponentially with score. • Raw Score: The score of an alignment, S, calculated as the sum of substitution and gap scores. Substitution scores are given by a look-up table (see PAM, BLOSUM). Gap scores are typically calculated as the sum of G, the gap opening penalty and L, the gap extension penalty. For a gap of length n, the gap cost would be G+Ln. The choice of gap costs, G and L is empirical, but it is customary to choose a high value for G (10-15)and a low value for L (1-2). • HSP: High-scoring segment pair. Local alignments with no gaps that achieve one of the top alignment scores in a given search. Sources: • http://www.ncbi.nlm.nih.gov/Education/BLASTinfo/similarity.html • http://www.ncbi.nlm.nih.gov/Education/BLASTinfo/information3.html
There are many metalloenzymes often doing crucial cellular biochemical (and biogeochemical) processes Enzymes containing metals: • Superoxide dismutase • Urease • Aconitase • Zinc finger proteins • Carbonic anhydrase • Alkaline phosphatase • DNA polymerase • Nitrate Reductase • Multi-copper oxidase • uvrA (ultraviolet resistence gene) • Ferredoxin • Nitrogenase • Many more… There are also many proteins and enzymes that are involved in metal processes (uptake, storage, insertion, transformations etc).
Integrated Microbial GenomicsJoint Genome Institute, Department of Energy The U.S. Department of Energy (DOE) Office of Science supports innovative, high-impact, peer-reviewed biological science to seek solutions to difficult DOE mission challenges. These challenges include finding alternative sources of energy, understanding biological carbon cycling as it relates to global climate change, and cleaning up environmental wastes. • Cleanup of toxic-waste sites worldwide. • Production of novel therapeutic and preventive agents and pathways. • Energy generation and development of renewable energy sources (e.g., methane and hydrogen). • Production of chemical catalysts, reagents, and enzymes to improve efficiency of industrial processes. • Management of environmental carbon dioxide, which is related to climate change. • Detection of disease-causing organisms and monitoring of the safety of food and water supplies. • Use of genetically altered bacteria as living sensors (biosensors) to detect harmful chemicals in soil, air, or water. • Understanding of specialized systems used by microbial cells to live in natural environments with other cells. • http://microbialgenomics.energy.gov/index.shtml
The Integrated Microbial Genomes (IMG) system serves as a community resource for comparative analysis and annotation of all publicly available genomes from three domains of life, in a uniquely integrated context. Go To: http://img.jgi.doe.gov/
IMG Carts • Carts are needed since IMG resets your session’s cache when you leave the site. • Carts are an easy way to save a list of: • Organisms (eg. all cyanobacteria) • Genes (i.e you have a list of genes that code for superoxide dismutase in 16 different organisms) • Functions (you have a list of the most popular metalloenzymes in the form of COG, Pfam, TigerFam, or EC#) • Saved as tab delimited text files
In Class Exercise on IMG:http://img.jgi.doe.gov • Load genomes • Go to “FIND GENOMES” • Click “VIEW PHYTOGENETICALLY” • Click “CLEAR ALL” to unselect all genomes • Click “ALL” after Cyanobacteria listings to select all Cyanobacterial genomes • Click “SAVE SELECTIONS” to choose only these selected Cyanobacterial genomes. Note at top now it should say 40 genomes selected. • Gene Search for Superoxide Dismutase, using “FIND GENES” function • By “GENE SEARCH”: type in superoxide dismutase and hit search. Note that this will only return genes that have been “annotated” as a superoxide dismutase by a previous computer or human annotator. Go ahead and grab a sequence for Synechococcus strain WH8102’s nickel superoxide dismutase, by clicking on the 474bp to the clipboard (highlight the area and hit control-C). Note that this is the DNA sequence. • Click the “FIND GENES” tab and then the “BLAST” tab: Paste in the nickel superoxide dismutase into open box. • Choose BLASTn for nucleotide (DNA) search • Set the cutoff value to 1e-2, (less stringent). • Note that the best hit is where you got the sequence from. • Repeat, but now with the amino acid sequence instead of the DNA sequence
Blast results Sequences producing significant alignments: (bits) E-Value 637000314.NC_005070 Synechococcus sp. WH 8102, complete genome. 940 0.0 637000310.NC_007516 Synechococcus sp. CC9605, complete genome. 389 e-105 639857006.NZ_AATZ01000003 Synechococcus sp. BL107, unfinished se... 311 3e-82 637000311.NC_007513 Synechococcus sp. CC9902, complete genome. 287 4e-75 637000309.NC_008319 Synechococcus sp. CC9311, complete genome. 208 3e-51 640069323.NC_008820 Prochlorococcus marinus str. MIT 9303, compl... 168 3e-39 637000211.NC_005071 Prochlorococcus marinus str. MIT 9313, compl... 153 2e-34 640963030.NZ_ABCS01000039 Plesiocystis pacifica SIR-1, unfinishe... 68 7e-09 637000213.NC_005042 Prochlorococcus marinus subsp. marinus str. ... 54 1e-04 641228501.NC_009976 Prochlorococcus marinus str. MIT 9211, compl... 48 0.007
The COG database: new developments in phylogenetic classification of proteins from complete genomesRoman L. Tatusov, Darren A. Natale, Igor V. Garkavtsev, Tatiana A. Tatusova, Uma T. Shankavaram, Bachoti S. Rao, Boris Kiryutin, Michael Y. Galperin, Natalie D. Fedorova, and Eugene V. KooninaNational Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA The database of Clusters of Orthologous Groups of proteins (COGs), which represents an attempt on a phylogenetic classification of the proteins encoded in complete genomes, currently consists of 2791 COGs including 45 350 proteins from 30 genomes of bacteria, archaea and the yeast Saccharomyces cerevisiae (http://www.ncbi.nlm.nih.gov/COG). In addition, a supplement to the COGs is available, in which proteins encoded in the genomes of two multicellular eukaryotes, the nematode Caenorhabditis elegans and the fruit fly Drosophila melanogaster, and shared with bacteria and/or archaea were included. The new features added to the COG database include information pages with structural and functional details on each COG and literature references, improvements of the COGNITOR program that is used to fit new proteins into the COGs, and classification of genomes and COGs constructed by using principal component analysis. Growth dynamics of the COG set with the increase of number of included genomes. The circles show the sequence of genome inclusion according to the actual order of sequencing, and the smooth line shows the mean of 106 random permutations of the genome order. The colored area indicates the range between the maximal and minimal value for each point (number of genomes) in 106 random permutations. Nucleic Acids Res. 2001 January 1; 29(1): 22–28.






















