Download
1 / 1
10 likes | 133 Views
Characterization of microbial communities in a fluidized-pellet-bed bioreactor by DGGE analysis. Introduction.

E N D
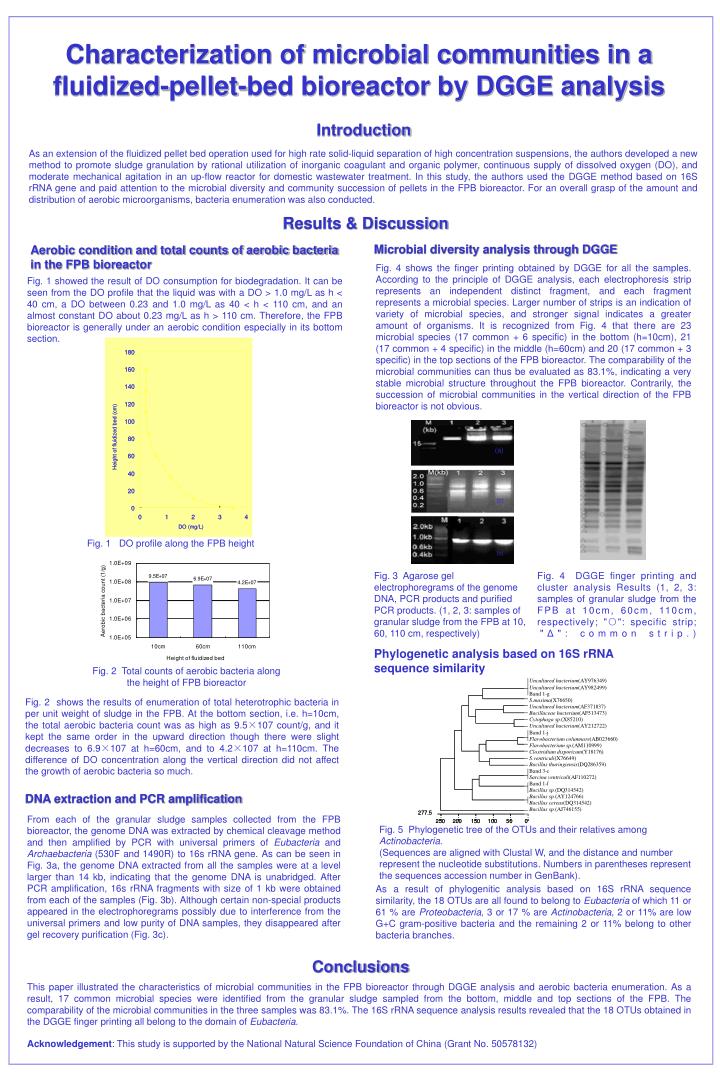
Characterization of microbial communities in a fluidized-pellet-bed bioreactor by DGGE analysis Introduction As an extension of the fluidized pellet bed operation used for high rate solid-liquid separation of high concentration suspensions, the authors developed a new method to promote sludge granulation by rational utilization of inorganic coagulant and organic polymer, continuous supply of dissolved oxygen (DO), and moderate mechanical agitation in an up-flow reactor for domestic wastewater treatment. In this study, the authors used the DGGE method based on 16S rRNA gene and paid attention to the microbial diversity and community succession of pellets in the FPB bioreactor. For an overall grasp of the amount and distribution of aerobic microorganisms, bacteria enumeration was also conducted. Results & Discussion Microbial diversity analysis through DGGE Aerobic condition and total counts of aerobic bacteria in the FPB bioreactor Fig. 4 shows the finger printing obtained by DGGE for all the samples. According to the principle of DGGE analysis, each electrophoresis strip represents an independent distinct fragment, and each fragment represents a microbial species. Larger number of strips is an indication of variety of microbial species, and stronger signal indicates a greater amount of organisms. It is recognized from Fig. 4 that there are 23 microbial species (17 common + 6 specific) in the bottom (h=10cm), 21 (17 common + 4 specific) in the middle (h=60cm) and 20 (17 common + 3 specific) in the top sections of the FPB bioreactor. The comparability of the microbial communities can thus be evaluated as 83.1%, indicating a very stable microbial structure throughout the FPB bioreactor. Contrarily, the succession of microbial communities in the vertical direction of the FPB bioreactor is not obvious. Fig. 1 showed the result of DO consumption for biodegradation. It can be seen from the DO profile that the liquid was with a DO > 1.0 mg/L as h < 40 cm, a DO between 0.23 and 1.0 mg/L as 40 < h < 110 cm, and an almost constant DO about 0.23 mg/L as h > 110 cm. Therefore, the FPB bioreactor is generally under an aerobic condition especially in its bottom section. (a) (b) Fig. 1 DO profile along the FPB height (c) Fig. 3 Agarose gel electrophoregrams of the genome DNA, PCR products and purified PCR products. (1, 2, 3: samples of granular sludge from the FPB at 10, 60, 110 cm, respectively) Fig. 4 DGGE finger printing and cluster analysis Results (1, 2, 3: samples of granular sludge from the FPB at 10cm, 60cm, 110cm, respectively; "○": specific strip; "Δ": common strip.) Phylogenetic analysis based on 16S rRNA sequence similarity Fig. 2 Total counts of aerobic bacteria along the height of FPB bioreactor Fig. 2 shows the results of enumeration of total heterotrophic bacteria in per unit weight of sludge in the FPB. At the bottom section, i.e. h=10cm, the total aerobic bacteria count was as high as 9.5×107 count/g, and it kept the same order in the upward direction though there were slight decreases to 6.9×107 at h=60cm, and to 4.2×107 at h=110cm. The difference of DO concentration along the vertical direction did not affect the growth of aerobic bacteria so much. DNA extraction and PCR amplification From each of the granular sludge samples collected from the FPB bioreactor, the genome DNA was extracted by chemical cleavage method and then amplified by PCR with universal primers of Eubacteria and Archaebacteria (530F and 1490R) to 16s rRNA gene. As can be seen in Fig. 3a, the genome DNA extracted from all the samples were at a level larger than 14 kb, indicating that the genome DNA is unabridged. After PCR amplification, 16s rRNA fragments with size of 1 kb were obtained from each of the samples (Fig. 3b). Although certain non-special products appeared in the electrophoregrams possibly due to interference from the universal primers and low purity of DNA samples, they disappeared after gel recovery purification (Fig. 3c). Fig. 5 Phylogenetic tree of the OTUs and their relatives among Actinobacteria. (Sequences are aligned with Clustal W, and the distance and number represent the nucleotide substitutions. Numbers in parentheses represent the sequences accession number in GenBank). As a result of phylogenitic analysis based on 16S rRNA sequence similarity, the 18 OTUs are all found to belong to Eubacteria of which 11 or 61 % are Proteobacteria, 3 or 17 % are Actinobacteria, 2 or 11% are low G+C gram-positive bacteria and the remaining 2 or 11% belong to other bacteria branches. Conclusions This paper illustrated the characteristics of microbial communities in the FPB bioreactor through DGGE analysis and aerobic bacteria enumeration. As a result, 17 common microbial species were identified from the granular sludge sampled from the bottom, middle and top sections of the FPB. The comparability of the microbial communities in the three samples was 83.1%. The 16S rRNA sequence analysis results revealed that the 18 OTUs obtained in the DGGE finger printing all belong to the domain of Eubacteria. Acknowledgement: This study is supported by the National Natural Science Foundation of China (Grant No. 50578132)