Download

1 / 29
530 likes | 2.1k Views
Restricted and Unrestricted Hartree-Fock method. Sudarshan Dhungana Phys790 Seminar (Feb15,2007). OVERVIEW. Introduction Approximations Hartree and Hartree-Fock methods Derivation of HF equations Basis functions Solution of RHF equation Results Advantages and Disadvantages References.

E N D
Restricted and Unrestricted Hartree-Fock method Sudarshan Dhungana Phys790 Seminar (Feb15,2007)
OVERVIEW • Introduction • Approximations • Hartree and Hartree-Fock methods • Derivation of HF equations • Basis functions • Solution of RHF equation • Results • Advantages and Disadvantages • References
Introduction • The HF method is widely used (mostly in the Quantum physics ,quantum chemistry community) because • By itself it can provide sufficiently accurate results, generally • In atoms and molecules • If we have the HF solution ,the accuracy can systematically be improved by applying various techniques(configuration interaction The HF method is based on Variational principle
Introduction • HF calculations with identical spatial orbitals for electrons with spins up/down are called restricted Hartree-Fock method • If different orbitals are used ,we have unrestricted Hartree-Fock method
Closed and open shell systems If the number of electrons is even and S=0,we have a closed shell (fig ’a’) • If the number of electrons is odd ,we have an open-shell system (fig .’b’) • In general ,if the numbers of electrons with spins up and down are different ,we have an open-shell system
Approximations • Born-Oppenheimer approach:the nuclei are fixed • The relativistic effects are completely neglected • The real Hamiltonian is essentilly replaced with a single –electron Hamiltonian
Approximations • The total WF can be represented as a single slater determinant but from the spin- orbitals
Difference between Hartree and Hartree Fock method Hartree-method: Multi electron wave function is approximated by a product of single –electron wave functions • Hartree-Fock method: Multi-electron wave function is approximated by a antisymmetrized sum of products of single –electron wave function
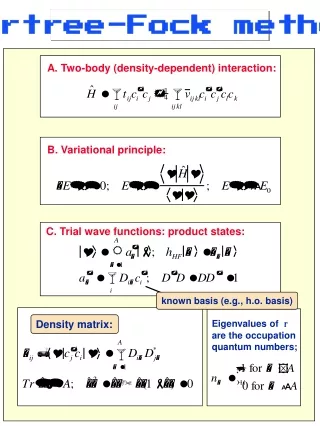
Derivation of the Hartree-Fock equation • The Hamiltonian reads: • where
Derivation of Hartree Fock equation • We write the total electron wave function as a Slater determinant built from orthonormal spin-orbitals • , • Note that if spin orbitals are normalized , is also normalized • In what follows ,the integrals means the integration over both spatial and sum over spin coordinates • Now we calculate the total energy as,
Derivation of Hartree Fock equation First part of total energy: • The second part of total energy: • Where
Derivation of Hartree Fock equation • Total energy: • We now define the Coulomb operator, • And exchange operator,
Derivation of Hartree Fock equation • In terms of these operators ,we can write the energy as • Now we determine the minimum of the functional as a function of the spin orbitals Ψk provided that they meet the orthonormality condition: • This implies that we have a minimisation problem with constraints,so we make use of the Lagrange multipliers • - =0
Derivation of Hartree Fock equation • Where, • Using the symmetry, • Introducing a new Hermitian Operator • (Fock operator)
Derivation of Hartree Fock equation • Because of the symmetry of the constraint equation ,we must have • Now : • And from the expression, - =0 • we arrive at:
Derivation of Hartree Fock equation • Since is small but arbitrary, which with leads to • The Lagrange parameter in the above equation can not be chosen freely: they must be such that the solutions form an orthonormal set .An obvious solution of the above equation is found by taking the as the eigen vectors of the Fock operators with eigen values and
Derivation of Hartree Fock equation • We have Schrödinger type equation • Can be called ‘orbitals’ • Can be called ‘orbital energies’ • The total energy • The Coulomb and exchange contributions must be subtracted off from the sum of Fock levels (to avoid counting the two –electron integrals twice)
Derivation of Hartree Fock equation • Some remarks • The sum here runs over all occupied levels • Eq. Is non linear, it must be solved by self-consistency procedure • Solving eq. Yields an infinite spectrum: to find the ground state ,we take the lowest N eigen states of the spectrum as the electron spin-orbitals. these orbitals are used to build a new Fock operator ,then the procedure is repeated over and over until the convergence is reached.
Solutions of Hartree fock equation • In order to solve the eigen value problem for the Fock operator, we have to expand the spin-orbitals in a basis set, that is we have to represent the orbitals as linear combinations of a finite no of basis states • Plane waves • perfect for crystals (periodic system) • Slater functions • Solutions for electrons in isolated atoms/small molecules • Gaussian functions
Solutions of Hartree fock equation • The general form of Fock operator is, • With • It is possible to eliminate the spin degrees of freedom by summing over them and find an operator acting only on spatial orbitals
Solutions of Hartree fock equation • The coulomb and exchange operators ,written in terms of orbital parts only read • The Fock operator now becomes, • Now for a given basis we obtain the following relation
Solutions of Hartree fock equation • The Fock matrix is now given by • Where, • Where k labels the orbitals and p ,q ,r label the basis functions • We introduce the density matrix, which (for RHF) is defined as
Solutions of Hartree Fock equation • The physical meaning of the Fock matrx is transparent :motion of an electron in the field of the nuclei and other electrons.Using density matrix Fock matrix is written as • And the total energy is
Unrestricted Hartree Fock Method • The spin up orbitals are represented by and spin down orbitals are represented by • With this vectors we reformulate HF equation • with
Application of restricted HF method(Closed shell atom) • For beryllium
Application of restricted HF method(open shell atom) • For Chlorine
References • Hartree Fock Program • Dr.Peter Winkler • Australian Journal of physics • Dr.J.Mitroy • Computational physics • Dr.J.M.Thijssen • Inroduction to electronic structure simulations • Dr.Arkady Krasheninnikov