Download

1 / 18
190 likes | 493 Views
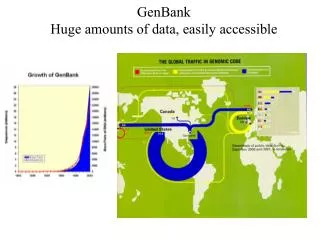
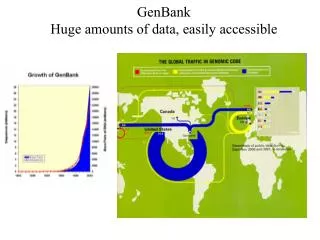
GenBank. Database di sequenze all’NIH 14,397,000,000 basi in 13,602,000 sequenze (Octobre 2001) Crescita esponenziale International Nucleotide Sequence Database Collaboration (DNA DataBank of Japan (DDBJ), European Molecular Biology Laboratory (EMBL), GenBank all NCBI)

E N D
GenBank Database di sequenze all’NIH 14,397,000,000 basi in 13,602,000 sequenze (Octobre 2001) Crescita esponenziale International Nucleotide Sequence Database Collaboration (DNA DataBank of Japan (DDBJ), European Molecular Biology Laboratory (EMBL), GenBank all NCBI) Scambio di informazioni ogni giorno
dbEST (sezione di GenBank) database pubblico di "Expressed Sequence Tags" (sequenze espresse contrassegnate), contiene tutte le sequenze ottenute dal sequenziamento parziale o totale di cloni di cDNA. dbEST release 102601 Number of public entries: 9,372,718 Summary by Organism - October 26, 2001 • Homo sapiens (human) 3,859,807 • Mus musculus + domesticus (mouse) 2,328,188 • Rattus sp. (rat) 317,076 • Drosophila melanogaster (fruit fly) 255,456 • ...
Sono disponibili le sequenze di entrambe le estremità del clone e le immagini dei cromatogrammi di sequenza possono essere facilmente raggiunte per controllarne la qualità. I cloni EST sono uno strumento molto utile per esperimenti di caratterizzazione di geni e studi di espressione. Una singola EST corrisponde ad un segmento molto breve rispetto all'estensione della regione codificante di un gene, ma essa costituisce il punto di partenza per la sua identificazione, che può essere ottenuta sia con i tradizionali sistemi di sequenziamento, che con metodi esclusivamente informatici (ad es. mediante "ESTassembly") che tentano di ricostruire la presumibile sequenza di consenso tra diverse EST parzialmente sovrapposte, identificandole nei database disponibili.
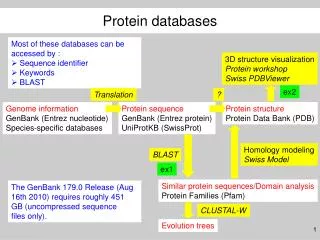
ENTREZ I Database: • Nucleotide • Protein • Genome • Structure • PopSet • Database Interlinking
Nucleotide Dati di sequenza da GenBank, EMBL, and DDBJ Protein Traduzione delle sequenze codificanti in GenBank, EMBL and DDBJ e sequenze di proteine sottomesse a PIR, SWISSPROT, PRF, Protein Data Bank (PDB) (sequenze da strutture risolte) Genome Sequenze di genomi completi di molti organismi Cromosomi completi Mappe di contigui Mappe genetiche/fisiche integrate
Structure Dati sperimentali di cristallografia e NMR Cn3D program PopSet Sequenze allineate, risultato di studi di genetica di popolazione, filogenesi e mutazione. Sia proteine che nucleotidi Database Interlinking
UniGene UniGene è il principale "gene indexing" database, mantenuto all'NCBI UniGene si propone di rappresentare l'insieme dei geni umani espressi attraverso il raggruppamento in un unico "cluster" di tutte le EST e le sequenze annotate di DNA genomico, mRNA, derivanti dai database GenBank e dbEST, simili tra loro e ipoteticamente afferenti alla medesima unità trascrizionale.
Il sistema di "clusterizzazione" si basa sulla similarità di sequenza e non sull'allineamento e le sequenze di scarsa qualità non vengono prese in considerazione. Le sequenze vengono comparate ognuna con tutte le altre in occasione di ciascuna delle frequenti versioni di UniGene e quelle che mostrano una similarità statisticamente significativa vengono inserite in un unico gruppo. Non viene costruita alcuna sequenza di consenso tra quelle di un "cluster", poiché a una singola unità trascrizionale possono corrispondere diversi contigui di sequenze espresse, a causa di fenomeni molto comuni quali o lo splicing alternativo o l'uso di diversi promotori per diverse isoforme.
Il processo di "clusterizzazione" si svolge in diversi passaggi, con stringenza decrescente. Prima vengono filtrate le sequenze contaminanti, ripetute o a bassa complessità e quelle ribosomiali e mitocondriali, in modo che ogni restante sequenza, di lunghezza superiore a 100 bp sia candidata per far parte di un "UniGene cluster". Poi vengono comparate tra loro e raggruppate le sequenze di geni e messaggeri; a questi "cluster" vengono aggiunte le EST correlate per similarità di sequenza o per informazioni sul clone di derivazione.
I "cluster" che non contengono il segnale di poliadenilazione vengono scartati, mantenendo solo i "cluster" "ancorati", ovvero quelli per cui è nota la sequenza 3', requisito fondamentale per l'identificazione di un gene. Gli ultimi stadi del processo provvedono all'assegnazione delle EST "orfane" e dei "cluster" di dimensione 1 a uno dei "cluster" "ancorati", con minore stringenza. Infine a ogni "cluster" viene assegnato il numero di identificazione, cercando di assicurare la massima continuità possibile con le precedenti versioni del database.
I parametri usati da UniGene per il processo di raggruppamento delle sequenze in "UniGene entry" sono caratterizzati da un grado di stringenza piuttosto basso percio’ ci si aspetta che esista in UniGene un singolo gruppo di trascritti a rappresentare ogni gene umano, ovvero che, di converso, le sequenze di trascritti diversi, ottenuti per splicing alternativo da un medesimo gene, siano raggruppate insieme in un'unica "entry" .
SAGE (Serial Analysis of Gene Expression) SAGE è un metodo sperimentale ideato per avere informazioni quantitative di espressione genica. SAGE consiste nel sequenziamento da messaggeri cellulari di brevi oligonucleotidi, che fungono da brevi etichette di sequenza (TAG) e si basa su tre principi: • - una sequenza di 9 paia di basi permette di identificare 49 (262144) diversi trascritti, dal momento che una "tag" viene ottenuta da una posizione specifica di ogni trascritto, • - le "tag" possono essere unite insieme in serie, a costituire lunghe molecole di DNA, che vengono clonate e sequenziate, • - il numero di volte in cui una singola "tag" viene osservata permette di quantificare l'abbondanza del messaggero identificato nella popolazione dei messaggeri e, indirettamente, il livello di espressione del gene corrispondente.
cDNA microarrays • Hundreds of thousands ESTs arranged in a single microscope slide by a robot • The basic principle is HYBRIDIZATION OF COMPLEMENTARY SEQUENCES • Determination of the level of activity of the genes represented by ESTs in the cell • Differential expression of genes in different samples(tissue type, normal/disease state, drugs effects)