Download
1 / 16
160 likes | 239 Views
IB404 - 18 - Human genome 4 – March 28.

E N D
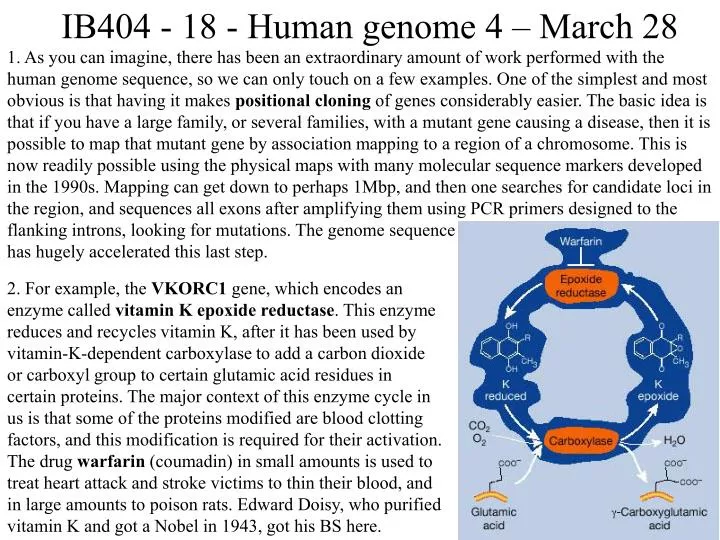
IB404 - 18 - Human genome 4 – March 28 1. As you can imagine, there has been an extraordinary amount of work performed with the human genome sequence, so we can only touch on a few examples. One of the simplest and most obvious is that having it makes positional cloning of genes considerably easier. The basic idea is that if you have a large family, or several families, with a mutant gene causing a disease, then it is possible to map that mutant gene by association mapping to a region of a chromosome. This is now readily possible using the physical maps with many molecular sequence markers developed in the 1990s. Mapping can get down to perhaps 1Mbp, and then one searches for candidate loci in the region, and sequences all exons after amplifying them using PCR primers designed to the flanking introns, looking for mutations. The genome sequence has hugely accelerated this last step. 2. For example, the VKORC1 gene, which encodes an enzyme called vitamin K epoxide reductase. This enzyme reduces and recycles vitamin K, after it has been used by vitamin-K-dependent carboxylase to add a carbon dioxide or carboxyl group to certain glutamic acid residues in certain proteins. The major context of this enzyme cycle in us is that some of the proteins modified are blood clotting factors, and this modification is required for their activation. The drug warfarin (coumadin) in small amounts is used to treat heart attack and stroke victims to thin their blood, and in large amounts to poison rats. Edward Doisy, who purified vitamin K and got a Nobel in 1943, got his BS here.
3. A German group mapped a mutation in a family with Combined Deficiency of Vitamin-K-Dependent Clotting Factors Type 2 to a 16 Mbp region of chromosome 16. In an effort to narrow the region down a little more they noted that there are a series of genes in this region that are orthologous and syntenic with a series of genes in the previously mapped warfarin resistance locus region in rats and mice. This narrowed the search to 4 Mbp, but there were still 130 annotated genes with ~1100 exons. 4. So they sequenced all ~1100 exons from all patients and parents in their families, plus in patients with resistance to warfarin treatment, plus in susceptible and resistant rats! The only gene that had mutations changing amino acids in all patients and resistant rats was a three-exon gene they call VKORC1, for vitamin K epoxide reductase complex subunit 1. In the homozygous patients with clotting disorders they found a single base change (C>T) causing an arginine to change to a tryptophan (arrows in top panel on next slide). Warfarin-resistant patients inherit this in a dominant fashion and in four patients they found four different heterozygous mutations, each again causing single amino acid changes, while the resistant rats were also heterozygous at yet another amino acid (“N” in bottom two panels). Thus amazingly all these mutations are simple single-base changes causing so-called missense mutations, that is, changes in single amino acids. You can imagine that many other kinds of mutations are possible, including frameshifts, stop codons (called nonsense mutations), small indels, splice junction mutations, promoter mutations, and others of no obvious effects, and these have been found in abundance in other genes, e.g. hundreds of mutations causing cystic fibrosis have been identified.
5. Human genes often have paralogs that were duplicated early in vertebrate evolution, often four of them (derived from two polyploidization events in early chordates), but ranging from none to many. VKORC1 turns out to have a single paralog in vertebrates, called VKORC1-Like1. In fact this turns out to be a more conservative protein evolutionarily, but its function is unknown. Presumably it has a related function because there is a single gene in other animals (except it has been lost from nematodes). I found a single gene in three protists, implying that this is an ancient gene, but lost from plants and fungi and other protists. There are also several retro- or processed pseudogenes in the human and rodent genomes for each paralog. 6. The public genome paper identified ±300 paralogs for ±1000 human disease genes, and these might be involved in related genetic diseases. Similarly, 18 novel paralogs of previous drug target genes were identified, which might then be new drug targets, e.g. another serotonin receptor subtype.
7. The molecular markers used for these kinds of mapping projects are called sequence tagged sites or STSs, and most commonly are microsatellites, that is, strings of repeats of di-, tri-, tetra- or penta-nucleotides where the repeat number varies. These are amplified using PCR primers designed to the flanking unique DNA, and length variants scored on a gel or chromatograph. This is also the technology used for forensic DNA work. Here is a dinucleotide microsatellite. Allele1 ACGGTCGATATGATAGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCG------------------TACCGCATATGTCATG Allele2 ACGGTCGATATGATAGCGCGCGCGCGCGCGCGCGCGCGCGCG------------------------TACCGCATATGTCATG Allele3 ACGGTCGATATGATAGCGCGCGCGCGCGCGCGCGCGCGCG--------------------------TACCGCATATGTCATG Allele4 ACGGTCGATATGATAGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGTACCGCATATGTCATG Primers ACGGTCGATATGATAG TACCGCATATGTCATG 8. More recently the sequencing of multiple human genomes has led to identification of millions of single nucleotide polymorphisms or SNPs, that is, places where human DNA commonly varies by a single base pair, roughly 1/1000bp or 3 million per genome copy. These can be typed or identified by various technologies and also used as genetic markers. One goal was to use these to map more complicated genetic traits, such as the basis for predisposition to heart attack or various cancers, that is, quantitative traits affected by alleles of several genes. It turns out this is hard to do technically and financially, so the HapMap project was undertaken to identify all the common haplotypes of multiple SNPs, hoping that this will simplify the requirement for typing of millions of SNPs. Of course, the difficulty is that haplotypes can be long or short, and for any particular region there could be many different haplotypes, all vestiges of our relatively young history as a species. And these are only the common ones, the assumption being that these common diseases will involve common haplotypes, contrasted with the rare single gene disorders like VKORC1 involving rare mutations.
9. Here are haplotypes of SNPs across a 100 kbp region in (A) and a 500 kbp region encoding 7 genes in (B). The SNPs were typed in ~400 individuals, and the resultant frequencies of haplotypes are shown in A. B shows the lengths of different haplotype blocks. Picking 2-3 SNPs to type for each haplotype block simplifies the required analysis down to less than 1m SNPs.
10. One of the promises of the human genome project was that it might eventually help reveal the genetic contribution to complex polygenic diseases like heart disease, diabetes, obesity, and various predispositions to cancers and other diseases. Because these are polygenic and hence difficult to map (except as QTLs or quantitative trait loci as is done in many species, but only identifies broad regions of the genome), there was a hope that typing or characterizing the SNP and haplotype patterns of sufficient numbers of patients might allow Genome Wide Association of SNP variants and haplotypes with susceptibility to disease (or reaction to drugs, known as pharmacogenomics; or other traits, like height, intelligence, or longevity). This is the promise of “personal genomics”, that by determining one’s SNP genotype one might obtain information about one’s own health. Ultimately, of course, complete diploid genomes would be the best, but it is still too expensive to do on the scale needed for GWA studies. To be statistically effective, given the huge numbers of SNPs being sampled, typically 500-600,000, representing all the major haplotype blocks in our genome, the numbers of patients and controls in these GWA studies needs to be in the thousands, preferably approaching 10,000. Given that each genotyping costs about $500, these are very expensive studies, but many have now been performed. This is why NIH is pushing so hard to get the cost of sequencing entire diploid genomes down to $1000, so GWAS can be repeated using entire genome sequences. Essentially one asks, is there a high probability of a particular SNP, usually between 1-10% of the alleles in the population, being associated with the disease or trait? There is no hypothesis, no candidate genes, it is a random undirected search of the entire genome with no preconceived ideas about what one might find.
11. Here is an example of a GWAS for metabolic traits, the kinds of measures doctors routinely make from blood tests these days, from the top, triglycerides, HDL, LDL, CRP, glucose, insulin, BMI, diastolic and systolic blood pressure. The dots each represent a SNP, with the chromosomes alternating in black and grey on each line. The Y axis is the log score indicating divergence from background, with the red line around log6 showing statistical significance. The vertical blue lines are regions previously associated with these traits. Some traits have many significant SNPs/regions (haplotypes), while others, like the bottom blood pressure tests, have none. Most earlier significant results were confirmed, but some are not. This is the kind of messiness one expects from tests in different populations.
12. While several hundred such studies have now been published since 2005, and many interesting associations of SNP haplotypes with diseases and traits have been identified, there are several major problems with these GWAS studies. One is that SNPs seldom contribute more than a few percent to the genetic variation for a trait, leaving a big question mark as to what the rest of the genetic heritability of these traits is determined by. In this metabolic study you can see the problem in this diagram. These SNPs only explain 2-20% of the known genetic variation. Two obvious possibilities is that it is synergistic combinations of SNPs and haplotypes that is important (epistasis), or alternatively alleles that are more rare than 1% in the population and have larger effects might be important, which will require whole genome sequencing to find. In addition, there are other kinds of genetic variation (four slides down) that might be important.
13. A second problem is that when the SNPs are examined in detail, they seldom are what you would hope, that is, they are seldom non-synonymous changes in exons of genes that encode proteins. Instead most are outside of genes, and at best identify regions of the genome of interest, with neighboring genes now being targeted for study. Nonetheless, they have given researchers hundreds of new leads in trying to identifying new genes involved in these many polygenic traits. 14. Some GWAS studies have not been successful, even though we know that the trait has a considerable genetic basis, most famously for “general intelligence” or g. The most recent study used the extremes of the range of g in ~10,000 children in the UK, and found only a few weak associations (blue dots), with none significant at log 6 (below).
15. Several GWAS have now attempted to determine the genetic contribution to long life. Clearly there is a major environmental component to long life as well, but surely there must be genetic contributions. One controversial study from Europe was just published. They compared 801 centenarians (median age at death of 104 years) with 914 reasonably matched controls. As we already know from many other studies including previous aging GWAS, the APOE gene has the most significant effect, with 100% prediction of aging (see Manhattan plot below). This apolipoprotein E has long been associated with Alzheimer’s disease, with several alleles predisposing, and others protecting. Having the protective alleles is essential to becoming a centenarian. But they extended this to a suite of 281 SNPs, which they call a “genetic model” of aging, which together have good predictive power for becoming a centenarian, and confirmed the model by repeating the analysis in two other cohorts of centenarians, including a group of 60 who lived to a mean age of 107 years. More refined analysis allowed separation or clustering of these SNPS into three separate groupings, suggesting three different ways to have a long-lived genetic inheritance. Some of the SNPs in other aging-associated genes are highlighted in the figure too.
16. For 5 years it has been possible to get yourself genotyped this way, by various “personal genomics” companies. The most famous is 23&Me, based in California, which for $300-500 will genotype ~500,000 SNPs from a spit sample you mail to them. Then you get access to all your raw data (the two nucleotides, A, C, G, T you have at each SNP position) on their website (useful for checking newly published GWAS), plus an interpretation of the results for traits, disease predisposition, and drug metabolism (primarily variants in p450 enzymes). These are all explained, with the relative importance of genetic and environmental factors made clear. I did this for my family, and the results are quite fascinating, even if not highly predictive, and as expected, not devastating. For some they have been quite instructive, for example Francis Collins discovered he has a predisposition for diabetes and made a radical lifestyle change as a result, while for others they can be devastating, for example Sergey Brin (co-founder of Google and husband of the co-founder of 23&Me, Anne Wojcicki), discovered that he is homozygous for a rare recessive allele that with almost certainty causes early onset Parkinson’s disease. The FDA does not approve of this kind of personal genetic testing, hence it has to be sold as “entertainment”, and there is a major debate in the research, medical, and ethics community about how readily available these kinds of genetic results should be. The doctors, of course, want to control it all, making even more money. I and many others think we should all be free to find out our own genetic inheritance, even if it means unpleasant discoveries, like your father is not really your biological father, or that you have a high likelihood of early Alzheimer’s disease. Some other companies doing personal genetic testing are Navigenics, Knome, and DecodeMe, although they have more restrictive policies on sharing your own data with you, for example, requiring that you consult with a doctor or genetic counselor, at considerably higher cost.
17. A related aspect of all this is that from the SNPs on your Y chromosome (for males) or your mitochondria (for females), your ancestry can be figured out with considerable resolution. This is because despite most genetic variation being between individuals, there are also residual differences in SNP frequencies between races and population groups and geographic regions. 23&Me will also provide these results, and even goes further today, identifying close and distant relatives who have also been genotyped by them, and allowing you to ask them to make contact with you (I’ve yet to ask any of my fourth and fifth cousins, or responded to the ~10 requests for contact I’ve had – they seem too distant to me - my close relatives are all in Scotland still). 18. This distance phylogenetic tree shows how this holds up across the entire world, for SNPs on all chromosomes. CEU is North Americans of European descent; CHB is Chinese, JPT is Japanese, YRI is Nigerians, and TGN and GDP are Tongans and Papua New Guineans (the study was confirming that Polynesians derived from SE Asia). Note that while most genetic divergence is indeed between individuals (the long lines to each individual colored dot), there is a common root to each group. The longer branch to YRI shows the divergence of the Out-of-Africa grouping.
19. But SNPs are only one kind of genetic variation between humans. The other major kind is called copy number variations or CNVs, and includes all insertions and deletions and duplications, from single bases to Mbp. It is much harder to assay all of these, and it was not until we had entire genomes from individuals that we realized that if you count up all the bases involved in these CNVs we differ by up to 0.5%, and perhaps more. From an evolutionary perspective, however, we count each of these CNVs as a single event. A GWAS in the UK of 19,000 individuals for 3,432 polymorphic CNVs longer than 500 bp was recently published, looking at eight diseases previously examined extensively by the same group using SNPs. They found just three CNVs associated with Crohn’s disease and diabetes, but all three regions had already been identified in their SNP GWAS, indicating that at least large CNVs are not responsible for the missing genetic heritability, at least for these diseases. 20. The same may not be true for a wide array of neurological problems, from schizophrenia to autism, where associations of large unique (non-inherited) CNVs are now being made. The strategy used to find them is quite remarkable. The researchers argued that these individuals are unlikely to reproduce effectively, hence their genetic defects might be unique (having occurred in one of their parents’ germlines), and large, affecting many genes. Indeed they found in many cases that individuals with major neurological defects have an excess of large CNVs versus controls, and most of these are unique and novel to them. Figuring out which genes in single or triple copies are the problem is a major task, something yet to be completely resolved even for Down Syndrome or trisomy-21, where an entire chromosome is present in three copies. Given that over 50% of genes are expressed in the brain, it is not surprising that having single or three copies of several genes might cause neurological problems.
21. Today most effort is directed towards sequencing entire diploid genomes. Venter did his using Sanger sequencing for ~$100m, James Watson was sequenced by 454 Life Sciences for ~$1m, then a series of Chinese, Nigerian, and others were sequenced with Illumina for $200,000 and dropping, now down to around $5,000. Meanwhile Complete Genomics with other re-sequencing methods claims to have it down to $3000, and two 1000-genome projects are underway here and in China. Eventually the idea is to get to $1000 per genome and do GWAS again with 10,000 patients and controls, this time hopefully finding the missing genetic variation! The bottom line from the comparisons of these genomes is that most non-Africans differ from each other by about 3 million SNPs and 300,000 CNVs, while African groups differ from each other, and everyone else, by around 4 million SNPs and about 400,000 CNVs. A recent paper described four different Khoisan men, and Bishop Desmond Tutu, revealing that indeed the San are the most divergent extant humans, even amongst themselves.
22. Finally, two years ago a group including Leroy Hood in Seattle finally did the obvious, and sequenced the genomes to two children with homozygous recessive genetic conditions that were previously unexplained, plus both parents - family-based sequencing. By eliminating all the common variants already known from the SNP and CNV databases, they were able to quickly narrow the search down to just a few candidate genes in which a child had inherited obvious potentially deleterious mutations from each parent, and quickly managed to identify the genetic basis for each condition. Furthermore they were able to show that each child inherited about 70 novel mutations from their parents, the first direct estimate of our actual mutation rate. They were also able to reveal the complete recombination pattern for every chromosome, showing 1-3 crossovers for short chromosomes and many for long ones, such as #4 below. The power gained by sequencing all four individuals was needed to get complete resolution here.





















