Download

1 / 58
650 likes | 978 Views
Introduction to Neurophysiology. Outline. Electrical Properties of Neurons Neuromuscular Junction Central Neurons Cerebral metabolism Autoregulation Elevated ICP Response to Injury . Electrical properties of neurons. Mechanism of Action Potential.

E N D
Outline • Electrical Properties of Neurons • Neuromuscular Junction • Central Neurons • Cerebral metabolism • Autoregulation • Elevated ICP • Response to Injury
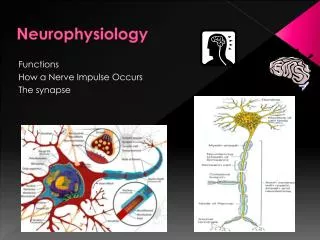
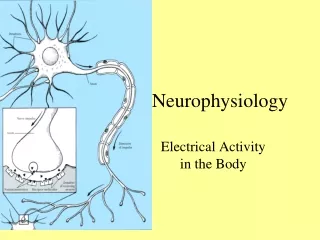
Mechanism of Action Potential • Membrane potential is caused by the separation of charge across the cell membrane (negative inside). • The action potential is generated by sequential opening of ion channels based on the membrane voltage. • Sodium and potassium gradients determine the membrane voltage. • At rest, there is a small passive flux of sodium into the cell and potassium out. • The Na/K/ATPase pump maintains the gradient.
Mechanism of the Action Potential • In response to a variety of stimuli, the membrane begins to depolarize. • Voltage sensitive Na channels open, allowing more Na inside. • The channels only remain open for a short time and then inactivated. • Meanwhile, the K channels open in response to the voltage change, K flows out and the membrane repolarizes. • The Na/K/ATPase pump is responsible for resetting the concentration gradients.
Mechanism of Propagation • The nature of the action potential allows for two important properties: summation and propagation. • Summation is the effect of multiple small depolarizations below threshold to add together to reach threshold. • Propagation of the action potential occurs when a local depolarization beyond threshold causes adjacent sodium channels to open sufficiently to cause depolarization. • The action potential can not spread backwards as the membrane is depolarized and needs to be reset first.
Neuromuscular Junction • Transmission between neurons or to muscles requires a conversion from electrical to chemical signals. • In general, the action potential propagates to an end terminal (aka pre-synaptic). • This causes Ca influx which causes the release of a chemical agent that floats across the synapse and binds with post-synaptic receptors.
Neuromuscular Junction • The receptors cause ion channels to open (or close) which changes the membrane conductance and depolarize the cell. • A variety of small molecules can act as a transmitter: • Ach, GABA, Glycine, Glutamate • Serotonin, Dopamine, Norepinephrine • However, the action of a transmitter does not depend on the nature of the chemical but on the properties of the receptor. • The receptor can be direct (transmitter binds directly to the ion channel) or indirect gated (receptor causes a cascade of second messengers that effect the ion channel).
Neuromuscular Junction • The muscle uses acetylcholine as its transmitter. • The transmitter (direct) gated channels in the muscle are permeable to both Na and K. • These channels are also not responsive to voltage which means that they are not regenerative. • The depolarization varies with the amount of transmitter released.
Central Neurons • Central neurons differ from motor neurons as they receive input from many presynaptic neurons, some can be inhibitory, and different neurotransmitters can have similar post-synaptic effects. • Each central neuron can be considered a summation machine. • Hundreds of axons synapse with hundreds of dendrites on each neuron. • Each synapse is typically unable to propagate an action potential through a cell body to the axon.
Central Neurons • This means that in order to transmit a signal, the neuron has to be stimulated by multiple synapses each opening a small number of Na/K channels (aka EPSP) until threshold is reached. • Inhibition of the action potential can be caused by neurotransmitters that open Cl channels; causing flow into the cell, making the inside more negative (aka IPSP) • If ∑EPSP - ∑IPSP > Threshold then AP = 1 • Else AP =0
Central Neurons • Glutamate is the major direct gated excitatory transmitter. • It binds to four different classes of receptors: • Kainate and Quisqualate A • AMPA • NMDA • NMDA is special because it opens Ca channels as well as Na and K and is voltage gated. • The Ca influx seems to act as a second messenger intracellularly.
Central Neurons • The major inhibitory transmitters are GABA and glycine. • These cause an increase in Cl conductance which hyperpolarizes the cell.
Central Neurons • Other neurotransmitters such as norepinephrine, acetylcholine and histamine act through a second messenger system. • Norepinephrine typically uses cAMP system. • Acetylcholine uses IP3/DAG/Ca/PKC systems. • Histamine uses the arachidonic acid pathways. • The major action is via ion channel phosphorylation that can either open or close the channel. • The second messengers can also affect gene expression.
Normal physiology of cerebral metabolism • Glucose is the sole energy substrate of the brain, unless there is ketosis. • Two terms are common in reference to metabolic turnover of glucose and oxygen: the cerebral metabolic rate of oxygen and the cerebral metabolic rate of glucose. • In awake adults the cerebral metabolic rate of oxygen is approximately 3.3 mg/100 g/min and the cerebral metabolic rate of glucose is 5.5 mg/100 g/min.
Of the total energy generated, 50% is used for interneuronal communication, generation, release, and uptake of neurotransmitters; 25% is used for maintenance and restoration of Ionic gradients across the cell membrane; and the remaining 25% is used for molecular transport, biosynthesis, and other as yet unidentified processes. • Most of the energy generated is consumed by neurons. • Glial cells that make up almost 50% of the brain have a much lower metabolic rate than neurons and account for less than 10% of total cerebral energy expenditure. • The brain accounts for only two to 3% of total body weight and does not do any mechanical work, yet it receives 20% of all cardiac output.
Regulation of cerebral blood flow • Substrate availability is determined by three factors: concentration of substrate in blood, flow volume, and rate of substrate passage across the blood brain barrier. • Usually the brain is able to maintain an adequate supply of substrates by regulation of cerebral blood flow. • Control of cerebral blood flow by adjustment in vessel diameter is common referred to as autoregulation of blood flow. • Several mechanisms active under different circumstances have been described.
Metabolic autoregulation • Cerebral blood flow is functionally coupled to cerebral metabolism, changing proportionally with increasing or decreasing regional or global metabolic demands. • Because 95% of the energy in the normal brain is generated by oxidative metabolism, cerebral metabolic rate of oxygen is considered a sensitive measure of cerebral metabolism. • In general, the brain responds to alterations in metabolism by changes in flow and thus has a tendency to keep AVDO2 relatively constant.
Pressure autoregulation • Changes in cerebral perfusion pressure will be followed by changes in cerebral blood flow unless diameter regulation takes place. • This type of autoregulation is termed pressure autoregulation and is the type of autoregulation referred to in most papers on autoregulation after head injury. • The limits of pressure autoregulation range from 40 to 150 mm of mercury of perfusion pressure. • Beyond these limits, vessel caliber follows flow passively leading to collapse of vessels at low pressure and forced dilatation or pressure breakthrough at high pressures.
Viscosity autoregulation • Cerebral blood flow can vary with changes in the viscosity of blood. • Increased viscosity increases cerebral vascular resistance. • This causes the vessels to dilate to decrease vascular resistance. • Thereby, cerebral blood flow is kept constant.
Theories on autoregulation • It is likely that a similar mechanism is involved in all three types of autoregulation. • Removal of the endothelium significantly reduces the contractions generated in response to various vasoconstrictors and hypoxia. • The observation that rapid elevation of transmural pressure triggers vasoconstriction and that this response is prevented by removal of the endothelium led to the idea that pressure on autoregulation may be endothelium mediated. • Two major endothelium derived contracting factors have been identified: thromboxane A2 and endothelin.
Carbon dioxide reactivity • Vascular caliber and cerebral blood flow are also responsive to changes in arterial CO2, a mechanism commonly referred to as CO2 reactivity. • Cerebral blood flow changes 2 to 3% for each mm Hg of CO2 between 20 to 60 mmHg. • Hypercarbia results in vasodilatation and higher cerebral blood flow, and hypocarbia results in vasoconstriction and lower cerebral blood flow.
Carbon Dioxide Reactivity • Vessels respond to pH in the perivascular space. • Over 24 hours the pH in the perivascular space and the diameter of cerebral blood vessels return to baseline. • With CO2 reactivity, changes in cerebral blood flow are compensated for by changes in AVDO2, so that a constant supply of substrates is maintained at the level set by metabolism. • This means that if a low CO2 causes vasoconstriction, the AVDO2 increases to compensate for the fall in delivery. • In contrast, a constant AVDO2 is a common feature of metabolic, pressure, and viscosity autoregulation.
Disturbance of Cerebral Metabolism • The cerebral metabolic rate of oxygen in comatose patients is typically reduced from a normal value of 3.2 mL per 100 g per minute to between 1.2 and 2.3 mL per 100 g per minute. • ATP generation from oxidative metabolism is impaired by either low supply (low cerebral blood flow or hypoxia) or dysfunctional processing (mitochondrial failure). • Cerebral acidosis occurs frequently after severe head injury. • Acidosis shifts the oxygen dissociation curve to the right, so that oxygen can be extracted more completely from hemoglobin. • Acidosis optimizes the pH for glycolysis and causes vasodilatation of blood vessels thus maximizing the available blood flow. • Functional recovery of tissue in the presence of high lactate is usually poor however.
Disturbances of Cerebral Blood Flow • Experimental data suggest that the brain becomes more vulnerable to ischemia after head injury. • Common metabolic and biochemical derangements or abnormal neurotransmitter receptor interactions, which reach a lethal threshold when the insults are combined, explain this increased vulnerability. • Under declining cerebral blood flow (due to failure of pressure autoregulation or severe hypotension), the brain can initially protect itself from ischemia and maintain metabolic supply by increasing extraction of the required oxygen from the flow. • Clinically this results in an increase AVDO2.
Disturbances of Cerebral Blood Flow: Impaired Vascular Reactivity • Metabolic autoregulation • In the presence of injury, blood flow increases. • It has been assumed that normal cerebral metabolism is depressed after severe head injury and this increased flow was luxury. • Cerebral blood flow is functionally coupled to cerebral metabolic rate of glucose. • Therefore increased cerebral blood flow is not necessarily luxury but may occur in response to increase glucose turnover or hyperglycolysis.
Disturbances of Cerebral Blood Flow: Impaired Vascular Reactivity • Pressure autoregulation • Autoregulation is usually temporarily dysfunctional for the first few days, with no apparent effect on outcome. • Based on clinical trials, it has been suggested that brainstem lesions damaging a brain stem autoregulatory center may be responsible. • Endothelial damage may also be responsible for the perturbation of pressure autoregulation. • Oxygen radicals that are generated after the initial injury may cause this endothelial damage.
Disturbances of Cerebral Blood Flow: Impaired Vascular Reactivity • Viscosity autoregulation • Cerebral blood flow increases in response to bolus administration of mannitol which reduces viscosity. • When pressure autoregulation is intact, the lower viscosity increases flow which causes the vessels to constrict and reduce total blood volume.
Disturbances of Cerebral Blood Flow: Impaired Vascular Reactivity • Carbon dioxide reactivity • CO2 reactivity is usually preserved after severe head injury. • It may be low early after injury, but in most cases, returns to a normal by 24 hours after injury. • Patients with severely impaired CO2 reactivity usually die or are left with severe neurological defects. • The pathological mechanisms leading to disturbed CO2 regulation are poorly understood. Free radicals may play a role.
Cerebral blood flow, cerebral blood volume, arteriovenous difference in oxygen, and autoregulation • Cerebral blood flow is influenced by vascular diameter, blood viscosity, and cerebral perfusion pressure, whereas cerebral blood volume is determined by vascular diameter only. • A decrease in cerebral metabolism results in a coupled decrease in cerebral blood flow obtained by vasoconstriction (metabolic autoregulation). • Cerebral blood volume decreases because of reduced vascular diameter, and AVDO2 remains constant because cerebral blood flow is tuned to metabolic demand.
Response to Failing Cerebral Perfusion Pressure • A reduction in cerebral perfusion pressure leads to compensatory vasodilatation with intact autoregulation. • Cerebral blood flow thus remains the same, but cerebral blood volume increases because of the larger vascular diameter. • Again, metabolism and cerebral blood flow are matched, and AVDO2 stays the same.
Response to Failing Cerebral Perfusion Pressure • Under certain circumstances, cerebral perfusion pressure decreases with defective autoregulation. • In this scenario, cerebral blood flow and cerebral blood volume follow the decrease in perfusion pressure passively. • AVDO2 increases, because cerebral blood flow is no longer adequate to meet the metabolic demand.
Response to Hyperventilation • Hypocapnia leads to a reduction in cerebral blood volume due to vasoconstriction. • Cerebral blood flow is also decreased and accompanied by increased oxygen extraction when cerebral blood flow is insufficient to meet the metabolic demand. • Hyperventilation causes a fall in ICP but at the cost of ischemia.
Raised intracranial pressure • By definition, intracranial pressure is the cerebral spinal fluid pressure. • Four parameters describe the static and dynamic CSF pressure: the rate of CSF production, the variable compliance given by the exponential relationship of CSF pressure to volume, the outflow resistance, and the intradural sinus pressure. • The Monro-Kellie doctrine states that the total volume of the intracranial contents (cerebral blood volume, CSF, and brain) is constant. • An increase in one of the three compartments must be accompanied by an equal decrease in one of the other compartments; otherwise ICP will increase.
As long as these volume compensations are sufficient, ICP remains relatively constant in the range of eight to 10 mmHg. • However, at a certain volume, this buffering capacity is exhausted, resulting in an exponential increase in pressure with any further volume addition. • The following five pathways of intracranial volume increases are: CSF system, cerebral blood volume, blood brain barrier damage associated edema (vasogenic edema), neurotoxic edema, and ischemic edema. • CSF components (CSF resistant to outflow and absorption) account for approximately one third of ICP elevation.
Cerebral blood flow is determined by the total diameter of the cerebral vascular bed. • Approximately 20 mL of blood (one third of the total cerebral blood volume) is located in the cerebral resistance vessels. • Because most autoregulatory and CO2 dependent diameter variations take place in these vessels, cerebral blood volume is determined mainly by their diameter.
Calcium • Trauma causes calcium to enter through voltage gated channels especially glutamate. • Excessive excitatory transmitters can cause cell death by: • Increased Na and Cl causing swelling • Increased intracellular Ca • Intracellular Ca attacks the cell membrane by activating PLA and PLC. • The fatty acids lead to breakdown of the BBB, cerebral edema, and generation of arachidonic acid by-products which affect blood flow.
Calcium • ROS are released by the mitochondria because of Ca mediated disruption of the ETC. • These ROS can cause DNA strand breaks.
Calpain and Cytoskeletal Proteolysis • Calpain are nonlysosomal cysteine proteases activated by increased intracellular Ca. • Activated calpains cause proteolysis of cytoskeletal proteins, neurofilament proteins and tubulin. • Calpains also contribute to the process that leads to cell death.
Mitochondria • Intramitochondrial Ca accumulation impairs the ETC, leading to less ATP and more ROS. • This causes cell death through energy failure and oxidative damage. • The increased Ca also causes increased permeability of the inner membrane leading to rupture.