Download

1 / 38
430 likes | 573 Views
Structural Biology. 8/27/10. Why determine structures?. Visualize primary sequence in context of folded protein (buried vs. solvent exposed). Highlight residues important for intermolecular interactions (co-crystals, packing, or computational (docking)).

E N D
Structural Biology 8/27/10
Why determine structures? Visualize primary sequence in context of folded protein (buried vs. solvent exposed) Highlight residues important for intermolecular interactions (co-crystals, packing, or computational (docking)) Visualize surface features to aid in identifying or designing binding partners (e.g. clefts, promontories, hydrophobic, or specifics of fold) Allow for the design of properly folded mutant proteins Allow use of structural databases to gain insight into function/evolution Provide a template for modeling studies to understand the function of related molecules
Structural Biology techniques ● Electron microscopy ≈ 5Å ? ● NMR equivalent resolution ≈ 2Å ● X-ray crystallography≈ 1Å ● Hybrid techniques EM + NMR/Crystallography
Resolution particulars Molecules secondary structure elements residues atoms 4.0 Å Low 3.0 Å Resolution 1.8 Å High 1.0 Å Center for Biological Sequence analysis DTU
X-ray crystallography & Nuclear Magnetic Resonance (NMR) X-ray crystallography utilizes information gleaned from bouncing X-rays off an ordered array of molecules. NMR utilizes information about magnetic environment of nuclei with non-zero spin NMR provides several snapshots of the object of interest all ~equally valid X-ray crystallography provides one snapshot of the object of interest. NMR cannot be practically used for large molecules (at least not yet). X-ray can be used for even very large molecules and complexes. Most importantly, structures that have been determined using both techniques are very similar!
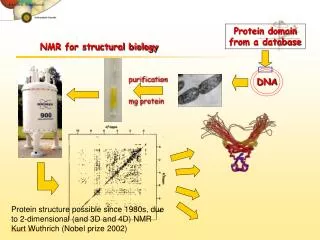
NMR (very basic drill) Purify protein Collect data Analyze Data (make assignments) Apply distance constraints and calculate structures
NMR - How it works NMR uses the behavior of nuclei with magnetic moments in an applied magnetic field. Biochemistry 5th edition,Berg, Tymoczko&Stryer For a given type of nucleus (1H), introduce RF radiation and excite transitions of nuclei from low to high energy state. Monitor emitted RF radiation as nuclei descend to low energy state (decay). FID (Free induction decay)
NMR (continued) De-convolute (separate) all the RF emissions from the FID to get a spectrum Individual nuclei transition at slightly different frequencies (resonances) depending on their chemical environments (electron clouds, other nuclei). The difference in resonance frequencies of nuclei from those of the same nuclei in a standard compound, are called chemical shifts. Therefore, Each protein has a unique spectrum for a given nuclei (1H, 13C, 15N,etc) Example reference compound trimethylsilane (TMS)
A nuclear Overhauser effect (NOE) experiment give peaks between protons that are close in space even though they’re not bonded. A correlation spectroscopy (COSY) experiment results in peaks between protons that are connected through covalent bonds. In this way, individual amino-acids have a characteristic signature (i.e. Ala vs. Ser). Intro to Protein Structure, Branden & Tooze By using COSY and NOESY experiments, one can identify various AAs and their neighboring AAs (sequential assignment). Once assignments are made, NOE info gives distance constraints. Distance constraints between atoms, once the atoms have been identified, reveal the structure!
Refinement is used in conjunction with known geometric and energetic constraints (in addition to the acquired distance constraints). Because of the limited number of distance constraints and the nature of solution-structure determination, one ends up with a set of structures that satisfy the distance criteria. So called “lowest penalty structures”. Kim et al.,Nature404, 151 - 158 (09 March 2000)
X-ray crystallography (basic drill) Grow crystals Collect diffraction data Solve structure
Protein phase diagram (constant temperature, pressure, pH) precipitate nucleation Clear metastable [precipitant] undersaturated [protein]
How do get a protein crystal? This is the hard part! Start with very pure protein Get a supersaturated solution Wait (sometimes a long time!) Keep trying….
Crystallization Most common technique is vapor diffusion. 0.3mm Drop(2L protein (20 mg/mL), 2L Reservoir solution) Reservoir(0.5 mL of 20% PEG 8,000, 200mM MgCl2, 100mM Tris pH 8) Cover with clear tape and place at RT or 4ºC Reservoir will slowly pull water out of drop and drop will concentrate. Hopefully you’ll get crystals. Many commercially available screens.
But remember, before you try crystallizing….. If you want to make a well-behaved, soluble expression construct spanning a region of a protein with unknown structure, you would: A) Do a data base search to identify other proteins with similar protein sequences • Use several different sequence alignment algorithms to align • any homologous sequences C) Use several different 2º structure prediction algorithms with your sequence of interest and any homologs D) Compare all of these 2º structure predictions (and decide!) E) Make several different constructs with different starts and stops F) All of the above Then you must work out expression, purification details!
When X-rays shine on atoms, the atoms become new sources of X-radiation. Each atom reflects X-rays in all directions. There is structural information in the “scattered X-rays, but it’s too weak when the atoms are from just one protein molecule. A crystal aligns a very large number of molecules in the same orientation. This provides the potential for a much stronger signal than when using just one molecule. Scattered X-rays reinforce in certain directions and cancel in most others X-rays crystal
Another way of thinking about it… Crystal is composed of many families of “planes” of atoms. Each family of planes are parallel and each is separated from the next by a specific distance “d”. Reflection of X-rays from these planes is reinforced when the geometric situation pictured above is achieved. Bragg’s law - 2dsinθ = n n usually = 1, is wavelength and is known
Two dimensional crystal “a” and “b” are the lengths of the sides of the unit cell (each unit cell in black). O is the origin. The sets of planes (green, blue, pink) are called Miller planes. The green set intersects the cell edge “a” at a=1/2 and cell edge “b” at b=1. Therefore, the green set of planes are the (2,1) of Miller planes. What you do is invert the 1/2 and it becomes 2. If the planes intersected “a” at 1/3, and “b” at 1/4, they would be the (3,4) family of Miller planes. Etc. You just look at the unit cell in the upper left corner – The planes are drawn in all the cells to show they intersect all the cells in the same way Note: if you slowly rotated this crystal in the X-ray beam, you would satisfy the requirements of Bragg’s law. Each set of planes would diffract in different directions.
This is a real diffraction pattern of a crystal in a special orientation (X-rays are being shined directly into the side of a unit cell) b a h k l Green (2,1,0) Blue (1,1,0) Pink (1,-1,0) Orange (-4,4,0) Every reflection arises from a different set of Miller planes. Every reflection has an index h,k,l – no two are the same.
This diffraction shows that this crystal has systematic absenses. But given the regularity of the diffraction pattern, we can easily measure the spacings along “a” and “b”. 1.5 mm Direct beam Where (1,0,0) would be So since we know the crystal to “film” distance, the wavelength, and where the spots are on the film, we can use geometry and calculate the size of “a” and “b”.
X-rays detector (1,0,0) 1.5 mm 80mm 2dsinθ = 1.5418Å (CuK) Re-arranging: d = 1.5418Å/2sinθ Solving: a = 82.2Å So 1.5/80 = tan 2θ tan 2θ = 0.01875 2θ = tan-1(0.01875) = 1.074º θ = 0.537º
We can do the same for b and c. Actually, programs have gotten so sophisticated, you feed any random orientation picture to a program and it scans the image, finds the spots and uses them to determine a,b,c, and any angles between them and the lattice type, the symmetry and the orientation of the crystal! In other words, the program knows the Miller indices for all the spots. So you simply start turning the crystal and collecting images. For example, you turn the crystal 1º and take a 1º oscillation picture. Do this for 180º, and you have a full data set. Integrate background (then subtract from spot) Integrate spot (Add counts in Pixels)
Do this for all (e.g.) ~40,000 spots in you data set. Now you must scale the spots from one image to the next (sometimes your shooting through a thicker part of the crystal etc.) When all spots have been integrated and scaled, you have a data set. Now each spot (h,k,l) should really be considered to be a wave. The intensity of the spot is the amplitude and the number of oscillations across the unit cell is revealed by its Miller indices. The (1,0,0) reflection would have one wavelength (of a sinusoidal wave) in the unit cell along the a direction, the (2,0,0) would be two wavecrests, etc. These waves can be added together – sometimes reinforcing, some- times cancelling out. When they’ve all been added together, they describe the shape of the “thing” that scattered them originally.
X-ray diffraction data Bragg’s Law: n = 2d sin Each data point has index and intensity h k l I 2 0 3 1483.6 3 -1 -3 19999.9 3 -1 -2 6729.6 3 -1 -1 30067.1 3 -1 1 8227.0 3 -1 2 29901.5 3 -1 3 24487.5 3 -1 4 502.1 3 dimensions Now all we need is the “phase” for each data point (reflection)
Fourier Series (1D example) f(x) = F0cos2(0x + 0) + F1cos2(1x + 1) + F2cos2(2x + 2) + F3cos2(3x + 3) + F4cos2(4x + 4) + . . . Fncos2(nx + n) f(x) = Fhcos2(hx + h) n h=1 Gale Rhodes Crystallography Made Crystal Clear(2nd edition)
The only trouble is, we must know the offset (phase) for each of the waves. In the previous 1D example, the phases were either 0º or 180º. Remember, we have ~40,000 of these “waves”. We know how tall they are and we know their wavelengths, but we don’t know the phases. The so-called “Phase Problem” 1 wavelength origin origin width of cell width of cell This ? Or this ?
One way to address this is to introduce a “heavy” atom into a crystal and collect another data set (say HG dataset). Now sinusoidal waves can also be represented as vectors. The length of the vector is the amplitude of the wave, the direction is the phase. = 45º 0, 360 Now we have two data sets. One set is HG the other is native (NAT). We can use a technique called the Patterson function to locate the coordinates of the Hg atom. The Patterson function doesn’t require phases. Once we locate (in x,y,z) the Hg atom, we actually know its contribution to each diffraction spot – its little vector!
Now Miller indices are a very convenient way of thinking about diffraction from a crystal. A more accurate way of thinking about what makes a given data point (h,k,l) relatively intense or weak is given by this formula: Fhkl is a vector. It is the sum of all the little vectors from all the atoms in the cell. But we have located the Hg atom so we know its x, y, z. So we know the direction and the phase for the contribution to the reflection made by the Hg atom! We will call this Fhg. Fhkl Fhg
So what we have are a bunch of |Fhkl|s – we have magnitudes but not directions. So we will represent them as circles with radii that are proportional to their magnitude. |FHG| |FNAT| Native reflection hkl HG reflection hkl And for the hkl reflection, we know vector Fhg (Note an Fhg for each hkl) We also know that |FNAT| + Fhg = |FHG| Or |FHG| - Fhg = |FNat|
Fhg |FHG| |FNAT| -Fhg Native HA |FHG| is offset by -Fhg -Fhg
Structure solved at CAMD IQGAP1 “GAP-related domain” 43kD