Download

1 / 26
260 likes | 309 Views
Additional Quantum-based Methods. a. Semi-empirical methods b. Density functional theory c. Molecular Orbital Applications. Semi-empirical methods. Extended Huckel MO Method (Wolfsberg, Helmholz, Hoffman) Independent electron approximation Schrodinger equation for electron i.

E N D
Additional Quantum-based Methods a. Semi-empirical methods b. Density functional theory c. Molecular Orbital Applications
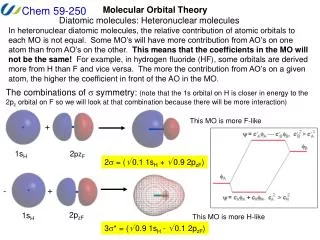
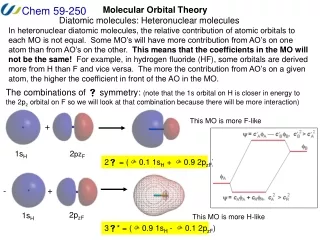
Extended Huckel MO Method (Wolfsberg, Helmholz, Hoffman) Independent electron approximation Schrodinger equation for electron i Hval = iHeff(i) Heff(i) = -(h2/2m) i2 + Veff(i) Heff(i) i = i i Semiempirical Molecular Orbital Calculation
LCAO-MO: fi= r criyr s (Heffrs- eiSrs ) csi = 0 Heffrs < r|Heff| s >Srs< r| s > • Parametrization: • Heffrr < r|Heff| r > • = minus the valence-state ionization • potential (VISP)
Atomic Orbital Energy VISP --------------- e5 -e5 --------------- e4 -e4 --------------- e3 -e3 --------------- e2 -e2 --------------- e1 -e1 Heffrs = ½ K(Heffrr + Heffss) SrsK: 13
CNDO, INDO, NDDO (Pople and co-workers) Hamiltonian with effective potentials Hval = i [ -(h2/2m) i2 + Veff(i) ] + ij>i e2 / rij two-electron integral: (rs|tu) = <r(1) t(2)| 1/r12 | s(1) u(2)> CNDO: complete neglect of differential overlap (rs|tu) = rs tu (rr|tt) rs tu rt
INDO: intermediate neglect of differential overlap (rs|tu) = 0 when r, s, t and u are not on the same atom. NDDO: neglect of diatomic differential overlap (rs|tu) = 0 if r and s (or t and u) are not on the same atom. CNDO, INDOare parametrized so that the overall results fit well with the results of minimal basis ab initio Hartree-Fock calculation. CNDO/S, INDO/S are parametrized to predict optical spectra.
MINDO, MNDO, AM1, PM3 (Dewar and co-workers, University of Texas, Austin) MINDO: modified INDO MNDO: modified neglect of diatomic overlap AM1: Austin Model 1 PM3: MNDO parametric method 3 *based on INDO & NDDO *reproduce the binding energy
Quantum Methods Electron Density Wavefunctions DFT Hartree-Fock TD-DFT MP2-CI The HF equations have to be solved iteratively because VHF depends upon solutions (the orbitals). In practice, one adopts the LCAO scheme, where the orbitals are expressed in terms of N basis functions, thus obtaining matricial equations depending upon N4bielectron integrals.
Information provided by is redundant N = 42e- benzene • Number of terms in the determinantal form : N! = 1.4 1051 • Number of Cartesian dimensions: 3N = 126 • is a very complex object including more information than we need! • The use of electron density allows to limit the redundant information • The electron density is a function of three coordinates no matter of the electron number.
What is Density? • Density provides us information about how something(s) is(are) distributed/spread about a given space • For a chemical system the electron density tells us where the electrons are likely to exist (e.g. allyl)
Representations of the electron density of the water molecule: (a) Relief map showing values of ρ(r) projected onto the plane, which contains the nuclei (large values near the oxygen atom are cut out); (b) Three dimensional molecular shape represented by an envelope of constant electron density (0.001 a.u.).
Definitions Function: a prescription which maps one or more numbers to another number: Functional: A functional takes a function as input and gives a number as output. An example is: Here f(x) is a function and y is a number. An example is the functional to integrate x from -to.
ab-initio methods can be interpreted as a functional of the wavefunction, with the functional form completely known! Can we write an explicit functional form of energy E[ρ]for DFT? In the general case the answer is not known. It is the main chalenge in DFT.
Timetable • 1920s: Introduction of the Thomas-Fermi model. • 1964: Hohenberg-Kohn paper proving existence of exact DF. • 1965: Kohn-Sham scheme introduced. • 1970s and early 80s: LDA. DFT becomes useful. • 1985: Incorporation of DFT into molecular dynamics (Car-Parrinello) (Now one of PRL’s top 10 cited papers). • 1988: Becke and LYP functionals. DFT useful for some chemistry. • 1998: Nobel prize awarded to Walter Kohn in chemistry for development of DFT.
Thomas-Fermi Energy (1920) -TF kinetic electron energy is an approximation of true kinetic electron energy -Potential nuclear-electron energy -Potential electron-electron Coulomb interaction energy exchange and correlation effects are completely neglected
Correlation energy Exchange correlation: Electrons with the same spin (ms) do not move independently as a consequence of the Pauli exclusion principle. = 0 if two electrons with the same spin occupy the same point in space, independently of their charge. HF theory treats exactly the exchange correlation generating a non local exchange correlation potential. Coulomb correlation: Electrons cannot move independently as a consequence of their Coulomb repulsion even though they are characterized by different spin (ms). HF theory completely neglects the Coulomb correlation thus generating, in principle, significant mistakes. Post HF treatments are often necessary.
The Slater exchange functional The predecessor to modern DFT is Slater’s Xa method. This method was formulated in 1951 as an approximate solution to the Hartree-Fock equations. In this method the HF exchange was approximated by: The exchange energy EXa is a fairly simple function of the electron density r. The adjustable parameter a was empirically determined for each atom in the periodic table. Typically a is between 0.7 and 0.8. For a free electron gas a = 2/3.
The VWN Correlation Functional In ab initio calculations of the Hartree-Fock type electron correlation is also not included. However, it can be included by inclusion of configuration interaction (CI). In DFT calculations the correlation functional plays this role. The Vosko-Wilk-Nusair (VWN) correlation function is often added to the Slater exchange function to make a combination exchange-correlation functional. Exc = Ex + Ec The nomenclature here is not standardized and the correlation functionals themselves are very complicated functions.
Correlation Energy: Is it important? N2 molecule: Corelation energy~ 0.5% of the electronic energy ~ 50% of the binding energy!
DFT energy EDFT = ENN + ET + Ev + Ecoul + Eexch + Ecorr ENN - nuclear-nuclear repulsion energy, Ev - nuclear-electron attraction energy , Ecoul -electron-electron Coulomb repulsion energy are the same as those used in Hartree-Fock theory. ET - kinetic energy of the electrons energy Eexch - electron-electron exchange energy Are different from those used in Hartree-Fock theory. Ecorr -correlation energy of electrons of different spin is not accounted for in Hartree-Fock theory. Due to these differences, the exchange energies calculated exactly in Hartree-Fock theory cannot be used in density functional theory.
The important advances for practical calculations are in electronic density-functional theory. • From Hohenberg and Kohn (1964) • Energy is a functional of electron density: E[r] • Ground-state only, but the exact r minimizes E[r] • Then Kohn and Sham (1965) • Variational equations for a “local” functional: • where Exc contains electron correlation:
Nonlocal effects, introduced in early 1990’s, have made DFT powerful. • Kohn and Sham had the local exchange functional: • Need “nonlocal” effects of gradient, • Current approach: Hybrid functionals • Combine Hartree-Fock Ex and DFT contributions Ex +Ec • Numerous proposed functionals and combinations • Axel Becke’s BLYP, B3LYP, BH&HLYP; Truhlar’s M06 • Give excellent structures and frequencies, poorer energies.
Hybrid functionalsThe basic idea behind the hybrid functionals is to mix exchange energies calculated in an exact (Hartree-Fock-like) manner with those obtained from DFT methods in order to improve performance. Frequently used methods are: B3LYP method. Becke-3-LYP (B3LYP) uses a different mixing scheme involving three mixing parameters:EXC = 0.2*EX(HF) + 0.8*EX(LSDA) + 0.72*DEX(B88) + 0.81*EC(LYP) + 0.19*EC(VWN)In this latter case, the B88 (Becke) gradient correction to the local LSDA exchange energies carries its own scaling factor of 0.72 and the LYP (Lee, Yang, and Parr) gradient correction to the local VWN correlation energies carries its own scaling factor of 0.81.The three scaling factors have been derived through fitting the parameters to a set of thermochemical data