Download
1 / 40
400 likes | 548 Views
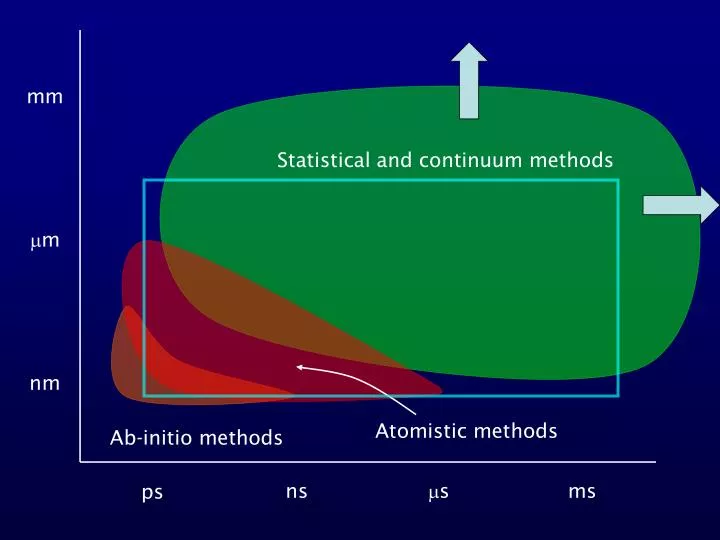
mm. m. nm. Atomistic methods. Ab-initio methods. ns. s. ms. ps. Statistical and continuum methods. What are atomistic simulations?. Simulations where atoms or groups of atoms are treated as classical particles. Cannot simulate chemical reactions.

E N D
mm m nm Atomistic methods Ab-initio methods ns s ms ps Statistical and continuum methods
What are atomistic simulations? • Simulations where atoms or groups of atoms are treated as classical particles. • Cannot simulate chemical reactions. • Electrostatic properties are approximated (electronic polarizability). • Other Approximations • The interaction potential is effective pair potential. • Boundary conditions. Simulation techniques • Molecular Dynamics (MD). • Monte Carlo (MC). • Brownian Dynamics (BD) etc. • Choice of the technique depends on the problem and the availability of computing resources.
Molecular dynamics algorithm : • Initialize positions, velocities and potential parameters • For each time step • Compute energy and forces • Update configuration • Non bonded forces • Bonded forces • External forces • Pressure tensor etc. • Compute new configuration by solving F = m a • Apply constraints and correct velocities • Scale simulation cell according to the ensemble
0.42 • Determination of partial charges on the atoms. • Get bond and angle parameters from spectroscopic data. • Work on smaller fragments of the molecule and determine LJ parameters by fitting the density and heat of vaporization. E.g. Linear chain alkanes were used to determine the LJ parameters of hydrocarbon chains in lipids. • Determination of dihedral parameters. • Again tune the parameters to match with known experimental details of the system. 0.42 -0.84 Force parameters
Bonded forces : • Usually static lists are enough and are easy to implement. • Parameter values depend on 1-4 non-bonded interactions.
Non bonded forces : • Lennard-Jones (LJ) potential • short range potential • can be truncated with a cutoff rc (errors??, dynamic neighbor list) • Electrostatics • long range potential • cutoffs usually produce erroneous results • Particle Mesh Ewald (PME) technique with appropriate boundary corrections are used. (poor scalability)
Membrane as a barrier Thanks to David Bostick
Fluid mosaic model Experiments are performed on model liposome vesicles
Fatty acid H e a d g r o u p G l y c e r o l Phosphate Hydrophilic Fatty acid Glycerophospholipid Hydrophobic lipid bilayer What is a lipid bilayer? Depending on the cross sectional area and the chain length the molecules can form vesicles, micelles, hexagonal phase
Key physical properties of lipid bilayers • Thickness. • increases with the length of the hydrocarbon chains • decreases with the number of unsaturated bond in chains • decreases with temperature • Area per molecule • increases with the length of the hydrocarbon chains • increases with the number of unsaturated bond in chains • increases with temperature • also depends on the properties of polar region
b Key physical properties of lipid bilayers • Order parameter
Bilayer exhibits first order phase transition with respect to temperature • The transition temperature Tm depends on • length of hydrocarbon chains • number of unsaturated bonds in the chain • properties of polar headgroup Key physical properties of lipid bilayers : Phase transition • Effect of phase transition • order parameters • thickness • repeat distance in multilamellar vesicles • area per lipid • bending and compressibility modulus. • lateral and transverse diffusion coefficients.
Lipid Area per lipid (Å2) simulations experiments Dipalmitoylphosphatidylcholine (DPPC) ~ 62 ~ 64 Dioleylphosphatidylcholine (DOPC) ~ 71 ~ 72 Dipalmitoylphosphatidylserine (DPPS with Na+ counter ions) ~ 53.6 ~ 54 Sphingomyelin (18:0) ~ 53 ~ 53 Cholesterol ~ 27 ~ 38* How real are the simulations ?
How real are the simulations ? Simulation of 128 DPPC bilayer in SPC water Pandit, Bostick and Berkowitz, Biophys. J. (2003), 84, 3743-3750
How real are the simulations ? Sometimes the comparison is not obvious e.g. Binding constants of various ions Binding constant of an ion is given by Langmuir isotherms where C is the concentration of ions at the membrane surface, is the ratio of number of bound ions to the number of lipids on the surface. Experimental values of binding constants of Na+ and Cl- to DPPC surface are KNa = 0.15 0.10 M-1 and KCl = 0.2 0.10 M-1
How real are the simulations ? KNa ~ 1.4 M-1 ?? KNa ~ 0.15 M-1 Pandit, Bostick and Berkowitz, Biophys. J. (2003), 84, 3743-3750
Water shells were observed using the surface to point correlation function. Predictability of atomistic simulations Pandit, Bostick and Berkowitz, J. Chem. Phys. (2003), 119(4), 2199-2205
b - face a - face Hydrophilic Heterogeneous Mixures in bilayers • Lipids with other membrane components. • Lipid mixtures with different headgroups and polar region (charged and zwitterionic lipids). • Lipid mixtures with different chain lengths and saturation. Mixture with cholesterol • Cholesterol increases mechanical strength of the bilayer • Ordering of chains and phase behavior.
Phase diagram with cholesterol • Lo or b phase • Liquid ordered phase where chain ordering is like gel phase but the lateral diffusion coefficients are like fluid phase. Reproduced from Vist and Davis (1990), Biochemistry, 29:451
Rafts • Rafts are liquid ordered domains in cell plasma membranes rich in sphingomyelin (or other sphingolipids), cholesterol and certain anchor proteins. • Rafts are important membrane structural components in: • signal transduction • protein transport • sorting of membrane components • process of fusion of bacteria and viruses into host cells How do rafts function and maintain their integrity? How does Cholesterol control the fluidity of lipid membranes?
2:1 mixture of POPC:cholesterol 2:1:1 mixture of POPC:cholesterol:SM 1:1:1 mixture of DOPC:SM:chol. Heterogeneous model membranes • Non-homogeneous domain formation is also observed in model membrane systems. Dietrich, C. et al, Biophys. J (2001), 80, 1417 • These domains are rich in sphingomyelin, cholesterol. • They are usually in liquid ordered or gel phase.
Simulation studies of rafts Study of ternary mixtures which form raft like domains. Study of liquid ordered phase Preformed domain Spontaneous domain formation • Mixture of cholesterol and saturated lipid (DPPC and DLPC). • Mixture of cholesterol and sphingomyelin. Ternary mixture of DOPC, SM, Chol with a preformed domain. Ternary mixture of DOPC, SM, Chol with random initial distribution
Study of Lo phase simulated systems: 120 PC molecules (DPPC/DLPC) 80 cholesterol molecules (~40 mol %) 5000 water molecules duration: 35 ns / 18 ns conditions: NPT ensemble (P = 1 atm, T = 323 / 279 => ) Pandit, Bostick and Berkowitz, Biophys. J. (2004), 86, 1345-1356.
DPPC+Cholesterol DLPC+Cholesterol Pandit, Bostick and Berkowitz, Biophys. J. (2004), 86, 1345-1356.
Conclusions from these simulations • Cholesterol increases order parameter of the lipids. The order parameters are close to liquid ordered phase order parameters. • Cholesterol forms complexes with two of its neighboring lipids. Life time of such a complex is ~10ns. • Complex formation process is dependent on the chain length of the lipids. Shorter chain lipids do not form large complexes. Pandit, Bostick and Berkowitz, Biophys. J. (2004), 86, 1345-1356.
Simulation of domain • 1424 DOPC, 266 SM, 122 cholesterols and 62561 waters (284,633 atoms) • Duration: 12 ns (with electrostatic cutoff) and 8 ns (with PME), 3 fs steps • Ensemble: NPT (P = 1 atm (isotropic), T = 293 K) • Configuration Biased Monte Carlo (CBMC) is used to thermalise chains. Pandit et al., Biophys. J. (2004),87,1092-1100.
Area per sphingomyelin: 49.5 Å2 /mol Area per DOPC: 61.0 Å2 /mol (~72 Å2 /mol) Area per Cholesterol: 29.6 Å2 /mol Pandit et al., Biophys. J. (2004),87,1092-1100.
Area/molecule of DOPC vs distance from SM-cholesterol domain DOPC molecular area affected by the ordered domain out to 8 nm Pandit et al., Biophys. J. (2004),87,1092-1100.
Early stages of nanoscopic domain formation? For the lipids in bilayer : D= 5 10 -12m2/s < r2 > = 4 D t t = 250 ns r ~ 22 Å In a system with 50 DOPC and 50 SM molecules < rSM-SM > ~ 9 Å If one can simulate a system for 100s of ns then one can probably see some of the processes involved in the early stages of domain formation Pandit, Jakobsson and Scott., Biophys. J. (2004), 87, 3312-3322.
0 ns 250 ns Binary mixture: 100 DOPC, 100 18:0 SM, 7000 water (31600 atoms) Pandit, Jakobsson and Scott., Biophys. J. (2004), 87, 3312-3322.
0 ns 250 ns Ternary mixture: 100 DOPC, 100 18:0 SM, 100 CHOL, 10000 water (43500 atoms) Pandit, Jakobsson and Scott., Biophys. J. (2004), 87, 3312-3322.
Body-centered axes for cholesterol: (x, y, z) Orientation of cholesterol Pandit, Jakobsson and Scott., Biophys. J. (2004), 87, 3312-3322.
- face of cholesterol SM packs well around the - face of cholesterol Pandit, Jakobsson and Scott., Biophys. J. (2004), 87, 3312-3322.
Speculation about structure of domain • Based on these simulations we speculate the structure of a ordered domain • To validate this speculation we need simulations that involve 103 to 105 lipids and simulation time scale of 10 s to 1 ms. This length and time scale is beyond the scope of current atomistic simulations.
Hybrid simulations with MD and MC • Combine MD with swapping moves of molecules. • MD simulation • Choose two molecules randomly and exchange their positions. • Place similar atoms of the molecules. • Place remaining atoms using CBMC. • Repeat step 2. for several pairs of molecules. • Go to step 1.
Coarse grain mapping 3D lipid bilayer 2D lattice field lipid chain Lattice field si Cholesterol Hard rod Khelashvili, Pandit and Scott., J. Chem. Phys. (2005), 123, 034910.
Binary mixture of DPPC and cholesterol Order parameter fields after 20 s of simulation with 10000 chains and 7%, 11%, 18%, 25% and 33% cholesterols. Khelashvili, Pandit and Scott., J. Chem. Phys. (2005), 123, 034910.
