Download
1 / 75
790 likes | 1.05k Views
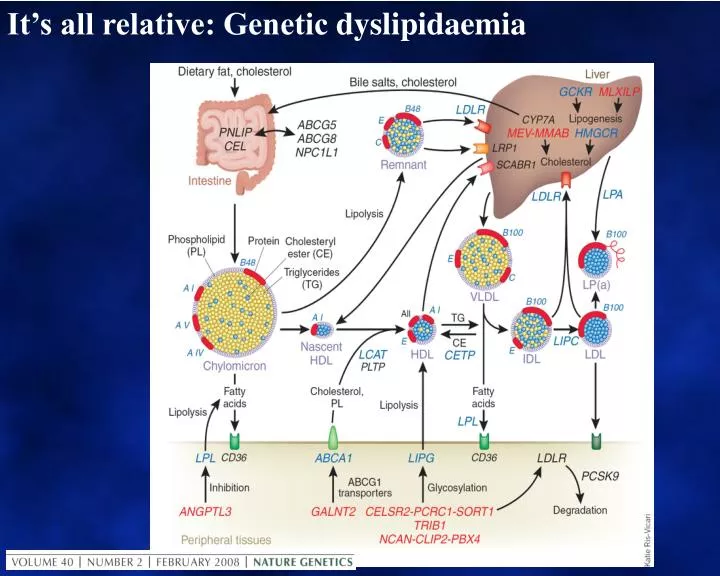
It’s all relative: Genetic dyslipidaemia. CVD risk factors, and especially lipid metabolism, exemplify gene / environment interactions. Mainly genetic Co-dominant mutations: Either genetic allele affected Recessive mutations: Both genetic alleles affected

E N D
CVD risk factors, and especially lipid metabolism, exemplify gene / environment interactions • Mainly genetic • Co-dominant mutations:Either genetic alleleaffected • Recessive mutations:Both genetic alleles affected • Polymorphisms and SNPs: Alleles consistent with “normal” and markers in proximity to significant genetic effects. Genome-wide association studies (GWAS). • Mainly environmental or secondary to other disorders.
A missing piece of the genetic puzzle Uncommon genes with large effect Pathogenic mutations Autosomal recessive Autosomal dominant Clinical Effect Polymorphisms Common genes with small effect Gene Prevalence
Genetic hyper and hypo - cholesterolaemia • Dominant Monogenic Hypercholesterolaemia • Familial hypercholesterolemia (FH) • Familial defective apo-100 (FDB-100) • PCSK9 gain-of-function (FH-3) • Hyperalphalipoproteinaemia (CETP deficiency, non-atherogenic?) • Recessive Monogenic Hypercholesterolaemia • Autosomal recessive hypercholesterolemia (ARH) • Lysosomal acid lipase deficiency: Wolman’s disease and Cholesterol ester storage disease • Dominant Monogenic LDL deficiency • PCSK9 loss-of-function • Recessive Monogenic LDL deficiency • Abeta and hypobeta – lipoproteinaemia • Chyomicron retention disease
CIII CIII CII CII AI AI . O AII MetabolicDefect in Familial Hypercholesterolemia B -100 INTESTINE VLDL LPL B -48 B E LPL LDL-R R B Chylomicrons E LIVER IDL LRP E FH SR-B1 E AI B HL HDL CE LDL LCAT B FC Macrophage OxLDL Other SR-B1 SR-A ABCA1 Tissues DAVIGNON 2006
CIII CIII CII CII SR-B1 E AI HDL CE AI . O LCAT AII FC Other SR-B1 Tissues MetabolicDefect in Familial DefectiveApoB-100 B Defective apoB B -100 INTESTINE VLDL LPL ApoB B -48 B E LPL LDL-R R B Chylomicrons E LIVER IDL LRP E FDB AI HL B LDL B Macrophage OxLDL SR-A ABCA1 DAVIGNON 2006
PCSK9 regulates the surface expression of LDLRs by targeting for lysosomal degradation 1. Qian YW, et al. J Lipid Res. 2007;48:1488-1498. 2. Horton JD, et al. J Lipid Res. 2009;50:S172-S177. 3. Zhang DW, et al. J Biol Chem. 2007;282:18602-18612.
Gain-of-Function Mutations in PCSK9 Cause Familial Hypercholesterolaemia (FH) 1. Abifadel M, et al. Hum Gen. 2009;30:520-529. 2. Horton JD, et al. J Lipid Res. 2009;50:S172-S177. 3. Cameron J, et al. Hum Mol Genet. 2006;15:1551-1558. • Associated with: • High serum LDL-C2 • Premature CHD and MI2 • In vitro testing in many identified mutations show decreased levels of LDLRs3 *For a full list of ADH mutations, please see refer to Abifadel reference.
FH is Co-dominant mutation of genes affecting formation or function of the LDL-receptor This causes metabolic and clinical consequences including precocious cardiovascular disease (CVD) Metabolic Increased LDL, Reduced clearance of remnants including LDL’s precursor, IDL. Increased Lp(a)? Reduced HDL? Clinical Dominant: 50% of each generation. Risk 50:50 Premature CHD, CVD and PVD Aortic stenosis Tendon xanthomas (11%) specific? Corneal arcus (27%) non-specific > 40y? Xanthelasmas (12%) nonspecific No signs highly sensitive FH IS NOT JUST HIGH CHOLESTEROL IN A PATIENT AND THEIR RELATIVE(S) What is “FH”? What does it cause?
FH: Why is it important? 1.0 Non-FH Women 0.9 Non-FH Men 0.8 FH Women 0.7 FH Men 0.6 Cumulative Probability of Clinical CAD 0.5 0.4 0.3 0.2 0.1 0.0 90+ 30-35 35-40 60-65 75-80 25-30 40-45 45-50 50-55 55-60 65-70 70-75 80-85 85-90 Age MED PED Registry 2001.
Prevalence: 0.2 – 0.5% (1 : 200-500) Atherosclerosis 173:55-68 > 8 x 106 affected world-wide. Seminars in Vasc Med 4:87-92 > 40,000 Australians Equal numbers of unaffected relatives Up to 1:60 in local groups with “founder effect”. Detection rates 0 – 44% World’s best 20-40% World average (including Australia) < 5% Impact: 5 – 10% of CHD events under age 60. J Lipid Res 34:269-77 CVD death in >80% of FH cases. Absolute CVD risk differs from general population models. High risk profile from birth, Interaction differs (smoking, gender) Circulation 97:1837-47 Risk algorithms underestimate risk Eur Heart J 19:A2-11 Missed and misdiagnosed Why FH matters: Prevalence and Impact Molecular Medicine meets Public Health
Diagnostic criteria Process Case detection: Dutch Lipid Clinic Score? Detecting index cases Optimal components Adults Diagnosis assessment Clinical services Children, Adolescents Model of Care for FH Laboratory protocol Genetic testing Management Clinical protocol Adults Cascade Screening Children, Adolescents Process LDL-Apheresis
Severe triglyceride elevation due to recessive impairment of lipoprotein lipase • Chylomicrons persist after fasting, massive levels of TG. • Homozygous deficiency of Lipoprotein Lipase (LPL) • Homozygous deficiency of the cofactor for L PL, Apo CII • Impaired transport of LPL to site of action (endothelium) due to homozygous defect in ANGPTL or GPIHBP • Combined overproduction and undercatabolism of triglyceride-rich lipoproteins sufficient to saturate LPL, eg in Apo AV mutations.
CIII CIII CII CII AI AI . O AII Metabolic Defects affecting Lipoprotein Lipase Dietary fat INTESTINE B -100 LPL B -48 VLDL B -48 E LDL-R TG CR B Chylomicrons E LPL LIVER IDL E LRP TG FFA + MG TG SR-B1 E AI B HL HDL CE LDL ApoB-48 R LCAT B FC VLDLR OxLDL Other SR-B1 Macrophage ABCA1 SR-A Tissues DAVIGNON 2006
Metabolic defects saturating in LPL, eg Apo AV mutations CIII CIII CII CII CETP AI TG CE AI . O AII ADIPOSE TISSUE Normal or reduced VLDL catabolism Sugar fat calories AV B -100 INTESTINE TG rich VLDL Overproduction of VLDL HSL LPL E B B -48 -48 B aGP + FFATG CR LPL IDL E E LDLR LRP SR-B1 Chylomicrons HL AV E B AI HDL sdLDL CE SR-A B LCAT FC VLDLR CD-36 OxLDL Macrophage Other LOX-1 SR-B1 ABCA1 Tissues SR-PSOX DAVIGNON 2006
Algorithm for Diagnosis of Apo B Dyslipoproteinemias HyperApo B > 1.2 g/L NormoApo B < 1.2 g/L NormoTG < 1.5 mmol/L NomoTG > 1.5 mmol/L Hyper TG > 1.5 mmol/L HyperTG > 1.5 mmol/L TG:Apo B >0.12 TG:Apo B < 0.12 Apo B > 0.75 g/L Apo B < 0.75 g/L TC:Apo B > 6.2 TC:Apo B < 6.2 Lipoproteins Normal Chylo + VLDL Chylo VLDL LDL VLDL + LDL Chylo + VLDL Remnants Primary Causes ■ Normal ■ Hypoalpha- lipoproteinemia ■ FCH ■ β-Sitosterolemia ■ Complete (FHC) or partial LPL deficiency associated with a secondary factor ■ Complete LPL deficiency (FHC) ■ Primary apoC-II deficiency ■ Familial dysbeta- lipoproteinemia (type III) ■ Hepatic lipase deficiency ■ (Primary cause associated with a secondary factor) ■ Familial hyperTG ■ Partial LPL deficiency ■ FH ■ Polygenic ■ FDB ■ PCSK9 deficiency ■ ARH deficiency ■ CYP7A1 deficiency ■ Hypoalphalipo-proteinemia Abbreviations: apo, apolipoprotein; ARH, autosomal recessive hypercholesterolemia; CAPD, continuous ambulatory peritoneal dialysis; Chylo, chylomicrons; CP7A1, cytochrome P450 7A1; DM2, diabetes mellitus type 2; dysbeta; dysbetalipoproteinemia; FCH, familial combined hyperlipidemia; FDB, familial defective apoB; FH, familial hypercholesterolemia; FHC, familial hyperchylomicronemia; HAART, highly active antiretroviral therapy; LPL, lipoprotein lipase; PCOS, polycystic ovary syndrome; SLE, systemic lupus erythematosus; TC, total cholesterol; TG, triglyceride. de Graaf J et al. Nat Clin Pract Endocrinol Metab 2008;4:608-18.
Autosomal recessive disorders have revealed HDL metabolism + Nascent HDL A-I A-I CE LCAT FC FC ABCA1 Macrophage Rapid catabolism Homozygous (?heterozygous) Apo AI deficiency: Atherogenic Tangier’s Disease: ABC-AI deficiency: Atherogenic? LCAT Deficiency: Non-atherogenic? Apo A1 Milano: Anti-atherogenic?
Carriers of the ApoAI Leu178Pro Variant Are at Increased Risk of Developing CAD 100 50 0 family controls (n=147) Event Free Survival ( % ) apoAI (L178P) carriers (n=54) p = 0.008 ApoAI 50% HDL-C 63% 18.9 x CAD risk 30 40 50 60 70 80 Age ( years) Hovingh K et al. J Amer Coll Cardiol 44:1429, 2004 DAVIGNON 2006
243 243 AIm/AIm 173 173 1 s s 1 Normal ApoAI and ApoAIMILANO Dimer Lipid Binding In Vivo Catabolism 35 143 187 99 243 AI 1 25 220 209 66 165 121 LCAT Activation Cholesterol Efflux “Receptor” Binding Franceschini G Eur J Clin Invest 26; 733, 1996 DAVIGNON 2006
AI C C E AII AI AI B 48 CII E OOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOO OOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOOO E Role of apolipoprotein E FC HDL CE • Apo E: ligand for hepatic removal of remnants. • Apo E knockout model is atherogenic • Apo E2 has lower binding affinity (E4>E3>E2). • E2:E2 only critical if lipids increase for other reasons. • Other roles for Apo E (CNS lipid transport and neural repair). TG TG LRP CHYLOMICRON LPL 48 B CIII E TG CR E Lipolysis products
Metabolic Defects in Remnant Dyslipidaemia Apo E2 homozygosity plus apo B overproduction or Hepatic Lipase deficiency AI AI AI E AI CE AII AI AI INTESTINE AII AII Degradation (catabolic) Formation (anabolic) E B FC/PL FC LPL PLTP Chylo to ChyloRe LCAT FC CE CE LIVER HL Kidney SR-B1 ABCA1 LRP LDLR E CE HDL3 HDL3 CELL B B LDL Ox LDL LCAT FC/PL HL E HL E TG CE B TG CE CETP VLDL to Remn CE HDL2 CE Modified from Deeb SS et al. J Lipid Res 44:1279, 2003
K IV Type 1 UnspecifiedhereditaryDyslipoproteinemia • Common: • Lipoptotein (a) • Rare: • Familial Phytosterolemia K IV Type 2 -N N LDL particle Apo(a) C ApoB LDL ReceptorBinding Site on apoB -C Protease K IV Types 3-10 K V Cholesterol ABCG5 ABCG8 Micelles Phytosterols DAVIGNON 2006 DAVIGNON 2006
Genetic dyslipidaemias have motivated treatment discovery • Clinical abnormalities represent real human problems • Massive yield on research into genetic dyslipidaemia • Familial Hypercholesterolaemia: Receptor mediated endocytosisStatins, PCSK9 antisense and Abs • Familial Hyperchylomicronaemia: LPL gene therapy • Apo A1 Milano: Synthetic HDL • Deficiency of Apo B or MTP MTP inhibitors, Apo B antisense • CETP Deficiency CETP inhibitors • Apo E Valuable animal k/o model
AAS MASTERCLASS 5-6TH OCTOBER 2012 IT’S ALL RELATIVE : MANAGEMENT OF GENETIC DYSLIPIDAEMIA CASE 1
HISTORY… Miss KM, 21year old Malay student nurse • Referred to Specialist Llipid Clinic • Hypercholesterolaemia • ‘Yellowish butterfly patterned lesion’ around her eyes • On Pravastatin 20mg ON • TC 13.7, LDL-c 11.6 mmol/L July 1997 • Cycling accident at her home town • Sustained minor soft tissue injury • Noted to have xanthelasma around her eyes • Lipid profile results: • TC : 15.4 mmol/L • LDL: 13.9 mmol/L • Referred to Medical Clinic, home town • Started Pravastatin 20 mg/ON 6th August 1998
HISTORY…… Noted ‘yellowish butterfly patterned lesion’ surrounding both eyes since primary school days Asymptomatic, well No h/o chest pain, palpitation, shortness of breath or poor effort tolerance No history of intermittent claudication or syncopal attacks. No h/o polyuria, polydipsia No h/o cold intolerance, lethargy or menstrual disturbance No past h/o jaundice, liver or renal diseases Not HT, DM No past surgical history
HISTORY… • The youngest out of 11 siblings • 3 siblings died : • eldest brother : died @ 43 years – AMI (HC) • elder brother (6th) : died @ 23 years – AMI (HC, xanthelasma) • eldest sister (2nd) : died @ 33 years - ?MI, ? SCD • Parents - consanguinous marriage (first cousins), not known to have DM/HPT/CAD
HISTORY… • Student nurse at a Teaching Hospital, KL • Single • Non smoker • No history of alcohol intake • Exercise - once weekly (jogging), 30minutes • Normal diet, low fiber intake
Physical Examination Anthropometry: BMI : 22.1 Waist circumference : 61cm (<80cm) Waist-to-hip ratio: 0.71 (<0.85) PR: 72/min, regular, BP : 120/62 mmHg, DRNM Peripheral pulses – present, carotid & renal bruit : absent Other systems: Normal
Corneal Arcus grading: 0 – no arcus observable 1- upper or lower segments affected 2- both segments affected 3- both segments and just confluent 4- heavy confluent arcus (Wider et al, 1983) Corneal Arcus
Digital xanthomata Achilles tendon xanthomata
Negative risk factors: Summary of KM’s risk factors Positive risk factors: Markedly elevated TC & LDL levels Strong family history of premature CAD HDL > 1.3 mmol/L Non smoker Not hypertensive Not DM Not overweight/ obese Premenopausal female
ECG – normal Exercise stress test – normal ECHO – Normal, no evidence of aortic stenosis Carotid artery IMT – normal, no evidence of atheromatous plaques Other Investigations….
Question 1 What are the various criteria for the diagnosis of FH? Dutch Lipid Clinic Network diagnostic scoring Simon Broome’s criteria MedPed criteria for FH NCEP ATPIII criteria
Question 2 According to the Dutch Lipid Clinic Network criteria, scoring for definite FH is: > 8 points 6 – 8 points 3 – 5 points
Question 3 In determining the Dutch Lipid Clinic diagnostic scoring for FH, the following are taken into account: Baseline LDL-c concentration: Yes/ No Clinical history of premature CAD: Yes/ No Clinical history of premature cerebral or PVD: Yes/ No Tendon xanthoma(ta) in the patient: Yes/ No Premature corneal arcus in the patient: Yes/ No Family history of hypercholesterolaemia: Yes/ No Family history of premature CAD/PVD in 1st degree relatives: Yes/ No Family history of tendon xanthomata and/or corneal arcus in 1st degree relatives: Yes/ No
Dutch Lipid Clinic – Diagnostic scoring for FH Criteria: • 8 points - DNA Mutation, or LDL-C > 8.5mmol/L • 6 points - Tendon xanthomas • 5 points - LDL-C 6.5 – 8.4mmol/L • 4 points - premature corneal arcus < 45 yrs • 3 points - LDL 5.0 – 6.4mmol/L • 2 points - 1st degree relative with xanthomas or premature CA or childhood LDL > 95th percentile, or personal premature CAD • 1 point - 1st deg relative with premature CAD/ vascular dis or LDL > 95th percentile, or personal history of premature cerebral or PVD, or LDL-c 4.0 – 4.9mmol/L Criteria: Family history: • 1st degree relative with (a) premature CAD or vascular dis (men<55yrs, women<60yrs) OR (b) LDL > 95th percentile, in - 1 point and / or • 1st degree relative with tendon xanthomata and/or corneal arcus OR childhood (<18yrs) LDL > 95th percentile - 2 points Clinical history • Patient with premature CAD (men<55, women <60yrs) - 2 points • Patient with premature cerebral or PVD (men<55, women <60yrs) -1 point Physical Examination - patient • Tendon xanthomas - 6 points • premature arcus - 4 points Lab Analysis – • LDL-c >8.5mmol/> - 8 points • LDL-c 6.5 – 8.4 - 5 • LDL-c 5.0 - 6.4 - 3 • LDL-c 4.0 – 4.9 - 1 DNA analysis • Functional DNA Mutation - 8 points Definite: > 8 points, Probable: 6 – 8 points, Possible FH: 3-5
US MedPed Criteria vs Simon Broome criteria for the dx of FH • Total cholesterol cutpoints (mmol/L) • 1st-degree vs 2nd-degree vs 3rd-degree relatives with FH vs General population • Age (years) 1st-degree 2nd degree 3rd degree General population • <20 5.7 5.9 6.2 7.0 • 20–29 6.2 6.5 6.7 7.5 • 30–39 7.0 7.2 7.5 8.8 • ≥40 7.5 7.8 8.0 9.3 • FH is diagnosed if TC levels exceed the cutpoint • Key: FH = familial hypercholesterolaemia
Simon Broome Criteria - Diagnosis of FH Criteria Description • (A) TC > 7.5 mmol/L in adults or TC > 6.7 mmol/L in children <16 years, or LDL-c > 4.9 mmol/L in adults or > 4.0 mmol/L in children • (B) Tendon xanthomas in the patient, or a first-degree or second-degree relative • (C ) DNA-based evidence of mutation in the LDLR, or apo- B100 or PCSK9 gene • (D) Family history of premature CHD (age <50 years in a second-degree relative or <60 years in a first-degree relative) • (E) Family history of raised TC >7.5 mmol/L in a first- or second-degree relative, or >6.7mmol/L in child, brother or sister <16yrs of age Diagnosis • Definite FH diagnosis requires either A + B or A + C • Possible FH diagnosis requires either A +D or A + E • Key: FH = familial hypercholesterolaemia
Summary Hypercholesterolaemia (LDL-c 13.9mmol/L) Xanthomata, corneal arcus, xanthelasma No evidence of personal CAD Strong family history of premature CAD Positive family history of HC – 1st and 2nd degree relative Consanguity Ruled out secondary causes of HC
CLINICAL DIAGNOSIS DEFINITE FAMILIAL HYPERCHOLESTROLEMIA
Question 4: Should risk estimation tools eg Framingham Risk Scoring be used for her? YES NO
CHD Risk Stratification CHD estimation tools such as those based on the Framingham risk scoring SHOULD NOT be used because people with FH are already at high risk of premature CHD. Heterozygous FH has >50% risk of CHD in men by the age of 50years, >30% in women by the age of 60years NICE Clinical Guideline 2008- Identification and Mx of FH 2.5.1 Individuals with FH are at high CHD risk. The 10-year CHD risk in the FH patient is not adequately predicted by any conventional risk assessment tools. Therefore, assessment of 10-year risk is not recommended.
Management • Statin therapy - continued, titrated • Add-on lipid lowering • Lifestyle modification • Referred to dietician • Referred to : - Cardiology - Plastic surgery • Family tracing, cascade screening and counselling