Download

1 / 49
490 likes | 658 Views
Introduction to Sequence Analysis Software & Data Libraries. Alex Ropelewski ropelews@psc.edu MARC: Developing Bioinformatics Programs July 17-28, 2006. Why Sequence Analysis.

E N D
Introduction to Sequence Analysis Software & Data Libraries Alex Ropelewski ropelews@psc.edu MARC: Developing Bioinformatics Programs July 17-28, 2006
Why Sequence Analysis • Sequence analysis is the process of applying computational methods to a biological sequence represented as a character string. • The goal is to use these computational methods to infer information about the structure, function, or evolutionary history of the sequence. • The stronger the evidence, the more confident we can be in the inference. • To get the strongest evidence the proper techniques must be employed. National Resource for Biomedical Supercomputing - An NIH Supported Research Center
CURATED DATASET: • All related sequences sharing a common function • All substantial motifs • Evolutionary history • Structural information • Experimental information The Goal National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Multiple Sequence Alignment Initial Query Profile & PSSM Sequence Libraries Local Patterns The Process Homology Modeling CURATED DATASET National Resource for Biomedical Supercomputing - An NIH Supported Research Center
The Project • Part I: Submit three candidate families for your course project. • Part II: Collect an initial set of sequences, generate a multiple sequence alignment • Part III: Improve the quality of your alignment, and identify additional family members • Part IV: Add structural and/or evolutionary information, and give a final report National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Multiple Sequence Alignment Initial Query Profile & PSSM Sequence Libraries Local Patterns Part I Homology Modeling CURATED DATASET National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Multiple Sequence Alignment Initial Query Profile & PSSM Local Patterns Part II Homology Modeling CURATED DATASET Classification Libraries Sequence Libraries National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Initial Query Part III Homology Modeling CURATED DATASET Multiple Sequence Alignment Sequence Libraries Local Patterns Profile & PSSM National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Hidden Markov Model Classification Libraries Initial Query Profile & PSSM Sequence Libraries Local Patterns Part IV Evolutionary Analysis Homology Modeling CURATED DATASET Multiple Sequence Alignment National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries • Structure libraries contain the actual three dimensional coordinates of a macromolecule. • Used to: • Determine if the three dimensional structure for a molecule has been solved • Visualize the three dimensional structure • Assist in homology modeling National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries • Protein Data Bank (PDB) • Large Molecules (1000+ atoms) • For more information see: • http://www.psc.edu/general/software/packages/pdb/pdb.html • http://www.rcsb.org/pdb/ • Cambridge Structural Database • Small Molecules (100+ atoms) • For more information see: • http://www.ccdc.cam.ac.uk/ National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Classification Libraries • Built from sets of related sequences and contain information about the residues that are essential to the structure/function of the group of related sequences • Used to: • Generate a testable hypothesis that the query sequence belongs to the group. • Quickly identify a good group of sequences known to share a biological relationship. National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Classification Libraries • Some Popular Classification Libraries: • PROSITE: http://www.expasy.ch/prosite.html • PFAM: http://pfam.wustl.edu/ • IPROCLASS: http://pir.georgetown.edu/iproclass/ • BLOCKS: http://www.blocks.fhcrc.org/ • PRINTS: http://www.biochem.ucl.ac.uk/bsm/dbbrowser/PRINTS/PRINTS.html • Transcription Factor Database: http://transfac.gbf.de/TRANSFAC/ • Restriction Enzyme Database: http://rebase.neb.com/ • Search software is usually specific to the database National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Classification Libraries - Representation • Consensus • Residue most common at each position in alignment • Composite • Set representation: (e.g. a {g,c} {acg} t a) • Composition Matrix • Table of how many residues present at each position • Position Specific Scoring Matrix (PSSM or Profile) • Log-odds likelihood of each residue at each position • Hidden Markov Model • Probabilistic state representation National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Sequence Libraries • Compilations of known sequences with experimental information about those sequences. • Used to: • Generate a testable hypothesis that the query sequence may be related to known sequences in the library. • Retrieve annotation information about sequences National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Sequence Libraries • Nucleic Acids • GenBank: http://www.ncbi.nlm.nih.gov/ • EMBL: http://www.ebi.ac.uk/ • Protein • UniProt/UniRef: http://www.uniprot.org/ • Other protein collections • PIR: http://nbrfa.georgetown.edu/ • Swiss-Prot: http://www.ebi.ac.uk/ • GenPept: http://www.ncbi.nlm.nih.gov/ • PIR-NREF: http://nbrfa.georgetown.edu/ • TREMBL: http://www.ebi.ac.uk/ • Older Libraries: PATCHX, OWL National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Sequence Libraries - Searching • Searching Methods: • Dynamic Programming: • Global: Needleman-Wunch, Sellers • Local: Smith-Waterman, Waterman-Egert "Maxsegs" • Approximations: • Fasta • Blast • User must understand what the searching method thinks similar means National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Sequence Libraries – Similar? Blood coagulation protein superfamily From The Molecular Basis of Blood Coagulation by Furie and Furie in Cell Vol 53 National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Sequence Libraries - Results Box 3.6 from Introduction to Bioinformatics by Attwood and Parry-Smith National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Multiple Sequence Alignment • An MSA is an alignment of a group of related sequences across their entire lengths in a manner than highlights the conservation of the important residues in the sequences • Critical building block for many next steps such as finding distantly related sequences, determining the evolutionary history, and homology modeling • Not all sets of related sequences can be aligned across their entire lengths cleanly National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Multiple Sequence Alignment Blood coagulation protein superfamily From The Molecular Basis of Blood Coagulation by Furie and Furie in Cell Vol 53 National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Multiple Sequence Alignment • When aligning groups of related sequences it is important to note that residues in those sequences are either: • Conserved (not mutated) • Unconstrained (when mutated can be almost any amino acid) • Constrained (when mutated must be one of a few amino acids) • Motifs are distinct units that consists of theconserved and constrained regions;Motifs generally tell us the residues that are essential to the structure/function of the sequence • Typically, multiple sequence alignments contain motifs as well as unconstrained regions. • Aligning motifs in a multiple sequence alignment will improve the quality of the alignment National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Multiple Sequence Alignment helix sheet helix Lrr 2ekdafrdlhsLsl-LsLydNnI-----qsL LRR A1 wteLlpllqqyEvvrLddCgLTeehCkdi LRR A2 lqgLqsPtCkiqkLsLqnCsLTeaGCgvL LRR A3 cegLldPqChLEkLqLeyCrLTaasCepL LRR A4 gqgLadsaCqLEtLrLenCgLTpanCkdL LRR A5 cpgLlsPasrLktLwLweCdiTasGCrdL LRR A6 cesLlqPGCQLEsLwvksCsLTaacCqhv LRR A7 cqaLsqPgttLrvLcLgdCeVTnsGCssL LRR A8 lgsLeQPgCaLEqLvLydtywTeevedrL LRR B1 gsaLranpsLtE-LcLrtNeLGDaGvhlv LRR B2 pstLrslptLrE-LhLsdNpLGDaGlrlL LRR B3 asvLratraLkE-LtvsnNdiGeaGarvL LRR B4 cgivasqasLrE-LDLgsNgLGDaGiaeL LRR B5 crvLqaketkKE-LsLagNkLGDeGarlL LRR B6 slmLtqnkhLlE-LqLssNkLGDsGiqeL LRR B7 aslLlanrsLRE-LdLsnNcvGDpGvlqL National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Multiple Sequence Alignment Figure 8.2 from Introduction to Bioinformatics by Attwood and Parry-Smith National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Multiple Sequence Alignment - Programs • MSA Using Progressive Pairwise Technique: • Clustal • MSA Using Multidimensional Dynamic Programming: • MSA • MSA Using Consistency Measures: • T-Coffee • Probcons • MSA Editor: • GeneDoc National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Position Specific Scoring Matrix • A Position Specific Scoring Matrix (PSSM or Profile) is a way to abstract the information contained in a multiple sequence alignment. • Think of a PSSM as a custom PAM or BLOSUM scoring matrix that has been specially tuned to locate sequences exactly like those in the alignment. • Probabilities represented by Log Odds Technique National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Position Specific Scoring Matrix • Used to: • Help locate distantly related sequences • Help resolve sequences that are not considered “statistically significant” by a database search, but share enough important residues to infer that the sequences may have the same function and be distant members of the sequence family • Good MSA = Good PSSM • Poor MSA = Poor PSSM • A lot of Sequences = Good PSSM • Few Sequences = Good PSSM National Resource for Biomedical Supercomputing - An NIH Supported Research Center
PSSM – Programs • MakePSSM • Used to create a PSSM from a multiple sequence alignment • PSSM can be created using different methods including Gribskov and Henikoff and with a variety of PAM and BLOSUM matrices. • ProfileSS • Used to search a sequence database with a profile. National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Hidden Markov Model • A Hidden Markov Model (HMM) is a way to abstract the information contained in a multiple sequence alignment. • Think of a HMM as a way to represent a multiple sequence alignment by deriving probabilities directly from the multiple sequence alignment • Probabilistic model – Includes probabilities for insertions and deletions National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Hidden Markov Model • Used to: • Help locate distantly related sequences • Help resolve sequences that are not considered “statistically significant” by a database search, but share enough important residues to infer that the sequences may have the same function and be distant members of the sequence family • Good MSA = Good HMM • Poor MSA = Poor HMM • A lot of Sequences = Good HMM • Few Sequences = Poor HMM National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Hidden Markov Model • HMMER Package: • hmmalign - Align multiple sequences to a profile HMM. • hmmbuild - Build a profile HMM from a given multiple sequencealignment. • hmmcalibrate - Determine appropriate statistical significance parameters for a profile HMM prior to doing database searches. • hmmconvert - Convert HMMER profile HMMs to other formats • hmmemit - Generate sequences probabilistically from a profile HMM. • hmmfetch - Retrieve an HMM from an HMM database • hmmindex - Create a binary SSI index for an HMM database • hmmpfam - Search a profile HMM database with a sequence hmmsearch - Search a sequence database with a profile HMM National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Local Patterns • Local patterns are short “motifs” that exist in all (or a subset) of the related sequences. • Finding local patterns may help us to align biologically important sections in a multiple sequence alignment. • These local patterns can also be used to probe sequence data libraries for distant relatives National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Local Patterns Blood coagulation protein superfamily From The Molecular Basis of Blood Coagulation by Furie and Furie in Cell Vol 53 National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Local Patterns - Programs • MEME – a tool for discovering motifs in groups of sequences • oops - One Occurrence Per Sequence • zoops - Zero or One Occurrence Per Sequence • tcm – Multiple occurrences per sequence • MAST – will search a sequence database for sequences that contain MEME patterns National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Homology Modeling • Predicts the three-dimensional structure of a given protein sequence (TARGET) based on an alignment to one or more known protein structures (TEMPLATES) • If similarity between the TARGET sequence and the TEMPLATE sequence is detected, structural similarity can be assumed. National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Homology Modeling Structural Superposition of Aldehyde Dehydrogenase Family Members National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Homology Modeling - Programs • Modeller – used for homology (or comparative) modeling of protein three-dimensional structures • MMTSB – Multiscale Modeling Tools for Structural Biology • VMD - molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting National Resource for Biomedical Supercomputing - An NIH Supported Research Center
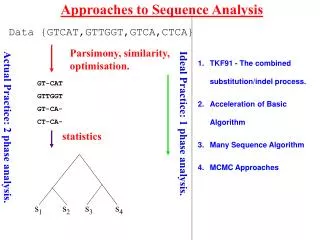
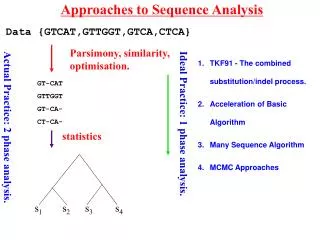
Evolutionary Analysis • Inferring phylogenies – finding the tree that implies the correct evolutionary history of the sequences • Principal Methods: • Parsimony analysis • Distance methods • Maximum Likelihood • Each approach has its own strengths/weaknesses • To assess the “correctness” of the tree we need to understand: • Overall signal & noise in the data • How the tree comparisons to alternate trees • How reliable the individual branches are National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Evolutionary Analysis • Refining Trees • Bootstrap analysis – can give us estimates of variability • Incorporating information about duplication and loss: • reconcile a gene tree with a species tree; • identify gene duplications • root an unrooted tree by minimizing gene duplications and losses; • refine rooted trees to minimize duplications and losses • Groups analysis • Discover what is unique about each subgroup in a tree • Help resolve which subgroup a sequence belongs to in a tree National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Evolutionary Analysis - Programs • Phylip Package: • Package contains many programs to help infer phylogenies including programs for bootstrapping, maximum parsimony, distance methods, maximal likelihood methods, etc. • Notung: • Enables the incorporation of information about duplication and loss into phylogenies • Subgroup Refinement: • GEnt – Calculates a group cross entropy National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Multiple Sequence Alignment Initial Query Profile & PSSM Sequence Libraries Local Patterns Workshop Project Homology Modeling CURATED DATASET National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Multiple Sequence Alignment Initial Query Profile & PSSM Sequence Libraries Local Patterns Part I Homology Modeling CURATED DATASET National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Workshop Project – Part I • Select Sequence Family: • We will be working with the Haloalkane Dehalogenase superfamily • A hydrolase that acts on halide bonds in C-halide compounds. • Reaction: 1-haloalkane + H(2)O = a primary alcohol + halide. • The PIR Superfamily is PIRSF037173 • The initial query sequence that we will be using for database searching is from Xanthobacter autrophicus. The UniProt ID is DHLA_XANAU. National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Multiple Sequence Alignment Initial Query Profile & PSSM Local Patterns Part II Homology Modeling CURATED DATASET Classification Libraries Sequence Libraries National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Workshop Project – Part II • Collect an initial set of sequences, generate a multiple sequence alignment • Perform a database search with the query sequence across several databases with different algorithms • Perform a multiple sequence alignment with a variety of different alignment algorithms • Select “best” multiple sequence alignment National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Evolutionary Analysis Hidden Markov Model Classification Libraries Initial Query Part III Homology Modeling CURATED DATASET Multiple Sequence Alignment Sequence Libraries Local Patterns Profile & PSSM National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Workshop Project – Part III • Improve the quality of your alignment, and identify additional family members • Search for local patterns in the group of sequences • Refine multiple sequence alignment based on local patterns • Convert alignment into HMM/PSSM and search the database for distantly related sequences. National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Structural Libraries Hidden Markov Model Classification Libraries Initial Query Profile & PSSM Sequence Libraries Local Patterns Part IV Evolutionary Analysis Homology Modeling CURATED DATASET Multiple Sequence Alignment National Resource for Biomedical Supercomputing - An NIH Supported Research Center
Workshop Project – Part IV • Add structural and evolutionary information • Build a phylogenetic tree • Refine the phylogenetic tree • Refine groups • Produce and visualize structure using homology modeling techniques • Produce final multiple sequence alignment National Resource for Biomedical Supercomputing - An NIH Supported Research Center