Download

1 / 55
550 likes | 806 Views
Clustering and Classification In Gene Expression Data. Carlo Colantuoni ccolantu@jhsph.edu. Slide Acknowledgements:

E N D
Clustering and Classification In Gene Expression Data Carlo Colantuoni ccolantu@jhsph.edu Slide Acknowledgements: Elizabeth Garrett-Mayer, Rafael Irizarry, Giovanni Parmigiani, David Madigan, Kevin Coombs, Richard Simon, Ingo Ruczinski.Classification based in part on Chapter 10 of Hand, Manilla, & Smyth and Chapter 7 of Han and Kamber
Data from Garber et al. PNAS (98), 2001.
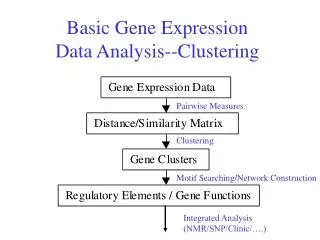
Clustering • Clustering is an exploratory tool to see who's running with who: Genes and Samples. • “Unsupervized” • NOT for classification of samples. • NOT for identification of differentially expressed genes.
Clustering • Clustering organizes things that are close into groups. • What does it mean for two genes to be close? • What does it mean for two samples to be close? • Once we know this, how do we define groups? • Hierarchical and K-Means Clustering
Distance • We need a mathematical definition of distance between two points • What are points? • If each gene is a point, what is the mathematical definition of a point?
Points 1 2 . . . . . . . N 1 2 . . . . . . . . G • Gene1= (E11, E12, …, E1N)’ • Gene2= (E21, E22, …, E2N)’ • Sample1= (E11, E21, …, EG1)’ • Sample2= (E12, E22, …, EG2)’ • Egi=expression gene g, sample i DATA MATRIX
Most Famous Distance • Euclidean distance • Example distance between gene 1 and 2: • Sqrt of Sum of (E1i -E2i)2, i=1,…,N • When N is 2, this is distance as we know it: Baltimore Distance DC When N is 20,000 you have to think abstractly
Correlation can also be used to compute distance • Pearson Correlation • Spearman Correlation • Uncentered Correlation • Absolute Value of Correlation
The difference is that, if you have two vectors X and Y with identical shape, but which are offset relative to each other by a fixed value, they will have a standard Pearson correlation (centered correlation) of 1 but will not have an uncentered correlation of 1.
The similarity/distance matrices 1 2 ………………………………...G 1 2 ……….N 1 2 . . . . . . . . G 1 2 . . . . . . . . G GENE SIMILARITY MATRIX DATA MATRIX
The similarity/distance matrices 1 2 ……….N 1 2 …………..N 1 2 . . . N 1 2 . . . . . . . . G SAMPLE SIMILARITY MATRIX DATA MATRIX
Gene and Sample Selection • Do you want all genes included? • What to do about replicates from the same individual/tumor? • Genes that contribute noise will affect your results. • Including all genes: dendrogram can’t all be seen at the same time. • Perhaps screen the genes?
Two commonly seen clustering approaches in gene expression data analysis • Hierarchical clustering • Dendrogram (red-green picture) • Allows us to cluster both genes and samples in one picture and see whole dataset “organized” • K-means/K-medoids • Partitioning method • Requires user to define K = # of clusters a priori • No picture to (over)interpret
Hierarchical Clustering • The most overused statistical method in gene expression analysis • Gives us pretty red-green picture with patterns • But, pretty picture tends to be pretty unstable. • Many different ways to perform hierarchical clustering • Tend to be sensitive to small changes in the data • Provided with clusters of every size: where to “cut” the dendrogram is user-determined
Choose clustering direction • Agglomerative clustering (bottom-up) • Starts with as each gene in its own cluster • Joins the two most similar clusters • Then, joins next two most similar clusters • Continues until all genes are in one cluster • Divisive clustering (top-down) • Starts with all genes in one cluster • Choose split so that genes in the two clusters are most similar (maximize “distance” between clusters) • Find next split in same manner • Continue until all genes are in single gene clusters
Choose linkage method (if bottom-up) • Single Linkage: join clusters whose distance between closest genes is smallest (elliptical) • Complete Linkage: join clusters whose distance between furthest genes is smallest (spherical) • Average Linkage: join clusters whose average distance is the smallest.
Simulated Data with 4 clusters: 1-10, 11-20, 21-30, 31-40 450 relevant genes + 450 “noise” genes. 450 relevant genes.
K-means and K-medoids • Partitioning Method • Don’t get pretty picture • MUST choose number of clusters K a priori • More of a “black box” because output is most commonly looked at purely as assignments • Each object (gene or sample) gets assigned to a cluster • Begin with initial partition • Iterate so that objects within clusters are most similar
K-means (continued) • Euclidean distance most often used • Spherical clusters. • Can be hard to choose or figure out K. • Not unique solution: clustering can depend on initial partition • No pretty figure to (over)interpret
K-means Algorithm 1. Choose K centroids at random 2. Make initial partition of objects into k clusters by assigning objects to closest centroid • Calculate the centroid (mean) of each of the k clusters. • a. For object i, calculate its distance to each of the centroids. b. Allocate object i to cluster with closest centroid. c. If object was reallocated, recalculate centroids based on new clusters. 4. Repeat 3 for object i = 1,….N. • Repeat 3 and 4 until no reallocations occur. • Assess cluster structure for fit and stability
We start with some data Interpretation: We are showing expression for two samples for 14 genes We are showing expression for two genes for 14 samples This is with 2 genes. K-means Iteration = 0
Choose K centroids These are starting values that the user picks. There are some data driven ways to do it K-means Iteration = 0
Make first partition by finding the closest centroid for each point This is where distance is used K-means Iteration = 1
Now re-compute the centroids by taking the middle of each cluster K-means Iteration = 2
Repeat until the centroids stop moving or until you get tired of waiting K-means Iteration = 3
K-means Limitations • Final results depend on starting values • How do we chose K? There are methods but not much theory saying what is best. • Where are the pretty pictures?
Assessing cluster fit and stability • Most often ignored. • Cluster structure is treated as reliable and precise • Can be VERY sensitive to noise and to outliers • Homogeneity and Separation • Cluster Silhouettes: how similar genes within a cluster are to genes in other clusters (Rousseeuw Journal of Computation and Applied Mathematics, 1987)
Silhouettes • Silhouette of gene i is defined as: • ai = average distance of gene i to other gene in same cluster • bi = average distance of gene i to genes in its nearest neighbor cluster
Add perturbations to original data Calculate the number of paired samples that cluster together in the original cluster that didn’t in the perturbed Repeat for every cutoff (i.e. for each k) Do iteratively Estimate for each k the proportion of discrepant pairs. WADP: Weighted Average Discrepancy Pairs
Classification • Diagnostic tests are good examples of classifiers • A patient has a given disease or not • The classifier is a machine that accepts some clinical parameters as input, and spits out an prediction for the patient • D • Not-D • Classes must be mutually exclusive and exhaustive
Components of Class Prediction • Select features (genes) • Which genes will be included in the model • Select type of classifier • E.g. (D)LDA, SVM, k-Nearest-Neighbor, … • Fit parameters for model (train the classifier) • Quantify predictive accuracy: Cross-Validation
Feature Selection • Goal is to identify a small subset of genes which together give accurate predictions. • Methods will vary depending on nature of classification problem • Choose genes with significant t-statistics to distinguish between two simple classes e.g.
Classifier Selection • In microarray classification, the number of features is (almost) always much greater than the number of samples. • Overfitting is a distinct risk, and increases with more complicated methods.
How microarrays differ from the rest of the world • Complex classification algorithms such as neural networks that perform better elsewhere don’t do as well as simpler methods for expression data. • Comparative studies have shown that simpler methods work as well or better for microarray problems because the number of candidate predictors exceeds the number of samples by orders of magnitude. (Dudoit, Fridlyand and Speed JASA 2001)
Statistical Methods Appropriate for Class Comparison may not be Appropriate for Class Prediction • Demonstrating statistical significance of prognostic factors is not the same as demonstrating predictive accuracy. • Demonstrating goodness of fit of a model to the data used to develop it is not a demonstration of predictive accuracy. • Most statistical methods were not developed for p>>n prediction problems
Linear discriminant analysis • If there are K classes, simply draw lines (planes) to divide the space of expression profiles into K regions, one for each class. • If profile X falls in region K, predict class K.
Nearest Neighbor Classification • To classify a new observation X, measure the distance d(X,Xi) between X and every sample Xi in training set • Assign to X the class label of its “nearest neighbor” in the training set.
Random Forests Build several “random” decision trees and have them vote to determine final classification
Evaluating a classifier • Want to estimate the error rate when classifier is used to predict class of a new observation • The ideal approach is to get a set of new observations, with known class label and see how frequently the classifier makes the correct prediction. • Performance on the training set is a poor approach, and will deflate the error estimate. • Cross validation methods are used to get less biased estimates of error using only the training data.
Split-Sample Evaluation • Training-set • Used to select features, select model type, determine parameters and cut-off thresholds • Test-set • Withheld until a single model is fully specified using the training-set. • Fully specified model is applied to the expression profiles in the test-set to predict class labels. • Number of errors is counted
V-fold cross validation • Divide data into V groups. • Hold one group back, train the classifier on other V-1 groups, and use it to predict the last one. • Rotate through all V points, holding each back. • Error estimate is total error rate on all V test groups.
Leave-one-out Cross Validation • Hold one data point back, train the classifier on other n-1 data points, and use it to predict the last one. • Rotate through all n points, holding each back. • Error estimate is total error rate on all n test values.
log-expression ratios full data set specimens log-expression ratios training set specimens test set Non-cross-validated Prediction 1. Prediction rule is built using full data set. 2. Rule is applied to each specimen for class prediction. Cross-validated Prediction (Leave-one-out method) 1. Full data set is divided into training and test sets (test set contains 1 specimen). 2. Prediction rule is built from scratch using the training set. 3. Rule is applied to the specimen in the test set for class prediction. 4. Process is repeated until each specimen has appeared once in the test set.
Which to use depends mostly on sample size • If the sample is large enough, split into test and train groups. • If sample is barely adequate for either testing or training, use leave one out • In between consider V-fold. This method can give more accurate estimates than leave one out, but reduces the size of training set.