Download
1 / 28
280 likes | 469 Views
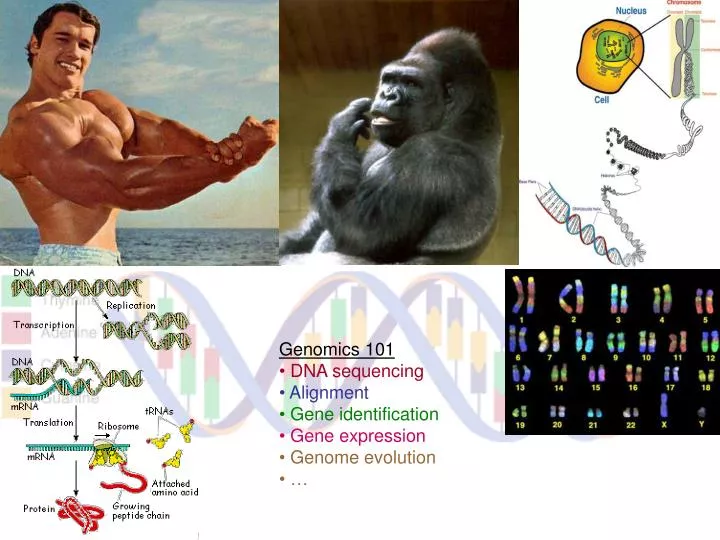
Genomics 101 DNA sequencing Alignment Gene identification Gene expression Genome evolution …. Next Few Topics. Gene Recognition Finding genes in DNA with computational methods Large-scale alignment & multiple alignment Comparing whole genomes, or large families of genes

E N D
Genomics 101 • DNA sequencing • Alignment • Gene identification • Gene expression • Genome evolution • …
Next Few Topics • Gene Recognition Finding genes in DNA with computational methods • Large-scale alignment & multiple alignment Comparing whole genomes, or large families of genes • Gene Expression and Regulation Measuring the expression of many genes at a time Finding elements in DNA that control the expression of genes
Gene Recognition Credits for slides: Marina Alexandersson Lior Pachter Serge Saxonov
Reading • GENSCAN • EasyGene • SLAM • Twinscan Optional: Chris Burge’s Thesis
DNA transcription RNA translation Protein Gene expression CCTGAGCCAACTATTGATGAA CCUGAGCCAACUAUUGAUGAA PEPTIDE
Gene structure intron1 intron2 exon2 exon3 exon1 transcription splicing translation Codon: A triplet of nucleotides that is converted to one amino acid exon = protein-coding intron = non-coding
In humans: ~22,000 genes ~1.5% of human DNA
Finding Genes • Exploit the regular gene structure ATG—Exon1—Intron1—Exon2—…—ExonN—STOP • Recognize “coding bias” CAG-CGA-GAC-TAT-TTA-GAT-AAC-ACA-CAT-GAA-… • Recognize splice sites Intron—cAGt—Exon—gGTgag—Intron • Model the duration of regions Introns tend to be much longer than exons, in mammals Exons are biased to have a given minimum length • Use cross-species comparison Gene structure is conserved in mammals Exons are more similar (~85%) than introns
Approaches to gene finding • Homology • BLAST, Procrustes. • Ab initio • Genscan, Genie, GeneID. • Hybrids • GenomeScan, GenieEST, Twinscan, SGP, ROSETTA, CEM, TBLASTX, SLAM.
Exon 3 Exon 1 Exon 2 Intron 1 Intron 2 5’ 3’ Stop codon TAG/TGA/TAA Start codon ATG 1. Exploit the regular gene structure Splice sites
Next Exon: Frame 0 Next Exon: Frame 1
2. Recognize “coding bias” • Each exon can be in one of three frames ag—gattacagattacagattaca—gtaag Frame 0 ag—gattacagattacagattaca—gtaag Frame 1 ag—gattacagattacagattaca—gtaag Frame 2 Frame of next exon depends on how many nucleotides are left over from previous exon • Codons “tag”, “tga”, and “taa” are STOP • No STOP codon appears in-frame, until end of gene • Absence of STOP is called open reading frame (ORF) • Different codons appear with different frequencies—codingbias
2. Recognize “coding bias” Amino Acid SLC DNA codons Isoleucine I ATT, ATC, ATA Leucine L CTT, CTC, CTA, CTG, TTA, TTG Valine V GTT, GTC, GTA, GTG Phenylalanine F TTT, TTC Methionine M ATG Cysteine C TGT, TGC Alanine A GCT, GCC, GCA, GCG Glycine G GGT, GGC, GGA, GGG Proline P CCT, CCC, CCA, CCG Threonine T ACT, ACC, ACA, ACG Serine S TCT, TCC, TCA, TCG, AGT, AGC Tyrosine Y TAT, TAC Tryptophan W TGG Glutamine Q CAA, CAG Asparagine N AAT, AAC Histidine H CAT, CAC Glutamic acid E GAA, GAG Aspartic acid D GAT, GAC Lysine K AAA, AAG Arginine R CGT, CGC, CGA, CGG, AGA, AGG Stop codons Stop TAA, TAG, TGA Can map 61 non-stop codons to frequencies & take log-odds ratios
atg caggtg ggtgag cagatg ggtgag cagttg ggtgag caggcc ggtgag tga
Biology of Splicing (http://genes.mit.edu/chris/)
3. Recognize splice sites Donor: 7.9 bits Acceptor: 9.4 bits (Stephens & Schneider, 1996) (http://www-lmmb.ncifcrf.gov/~toms/sequencelogo.html)
Donor site 5’ 3’ Position % 3. Recognize splice sites
3. Recognize splice sites • WMM: weight matrix model = PSSM (Staden 1984) • WAM: weight array model = 1st order Markov (Zhang & Marr 1993) • MDD: maximal dependence decomposition (Burge & Karlin 1997) • Decision-tree algorithm to take pairwise dependencies into account • For each position I, calculate Si = ji2(Ci, Xj) • Choose i* such that Si* is maximal and partition into two subsets, until • No significant dependencies left, or • Not enough sequences in subset • Train separate WMM models for each subset G5G-1 G5G-1 A2 G5G-1 A2U6 G5 All donor splice sites not G5 G5 not G-1 G5G-1 not A2 G5G-1A2 not U6
intron exon exon intron intergene exon intergene Hidden Markov Models for Gene Finding First Exon State Intron State Intergene State GTCAGATGAGCAAAGTAGACACTCCAGTAACGCGGTGAGTACATTAA
intron exon exon intron intergene exon intergene Hidden Markov Models for Gene Finding First Exon State Intron State Intergene State GTCAGATGAGCAAAGTAGACACTCCAGTAACGCGGTGAGTACATTAA
T A A T A T G T C C A C G G G T A T T G A G C A T T G T A C A C G G G G T A T T G A G C A T G T A A T G A A Exon1 Exon2 Exon3 Duration HMM for Gene Finding Duration Modeling Introns: regular HMM states—geometric duration Exons: special duration model VE0,0(i) = maxd=1…D { Prob[duration(E0,0)=d]aIntron0,E0,0 j=i-d+1…ieE0,0(xj) } where i is an admissible exon-ending state, D is restricted by the longest ORF GENSCAN: Chris Burge and Sam Karlin, 1997 Best performing de novo gene finder HMM with duration modeling for Exon states duration
HMM-based Gene Finders • GENSCAN (Burge 1997) • Big jump in accuracy of de novo gene finding • Currently, one of the best • HMM with duration modeling for Exon states • FGENESH (Solovyev 1997) • Currently one of the best • HMMgene (Krogh 1997) • GENIE (Kulp 1996) • GENMARK (Borodovsky & McIninch 1993) • VEIL (Henderson, Salzberg, & Fasman 1997)
Better way to do it: negative binomial • EasyGene: Prokaryotic gene-finder Larsen TS, Krogh A • Negative binomial with n = 3
GENSCAN’s hidden weapon • C+G content is correlated with: • Gene content (+) • Mean exon length (+) • Mean intron length (–) • These quantities affect parameters of model • Solution • Train parameters of model in four different C+G content ranges!
TP FP TN FN TP FN TN Actual Predicted Actual TP FP Predicted No Coding / Coding FN TN Evaluation of Accuracy Coding / No Coding (Slide by NF Samatova)
Results of GENSCAN • On the initial test dataset (Burset & Guigo) • 80% exact exon detection • 10% partial exons • 10% wrong exons • In general • HMMs have been best in de novo prediction • In practice they overpredict human genes by ~2x











![[III] Genes, Genomics, and Chromosomes](https://cdn1.slideserve.com/3548265/iii-genes-genomics-and-chromosomes-dt.jpg)








