Download
1 / 35
350 likes | 448 Views
Genome-wide Association S tudy. Focus on association between SNPs and traits Tendency Larger and larger sample size Use of more narrowly defined phenotypes(blood lipids, proinsulin or similar biomarkers Limitations Sufficient sample size

E N D
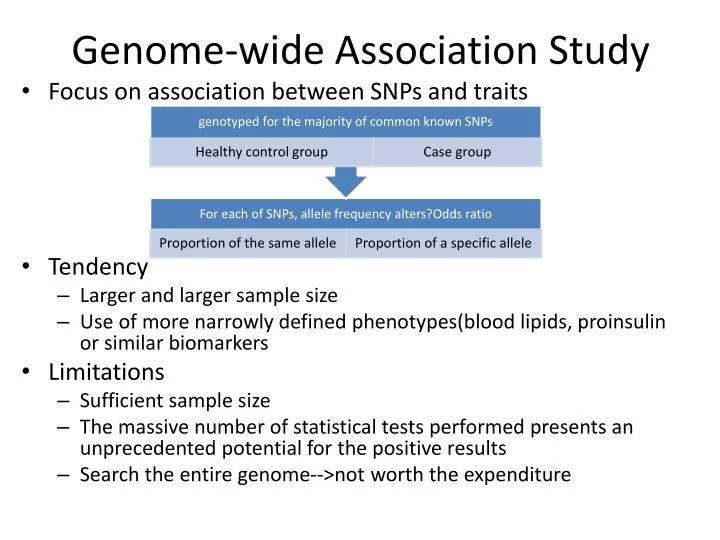
Genome-wide Association Study • Focus on association between SNPs and traits • Tendency • Larger and larger sample size • Use of more narrowly defined phenotypes(blood lipids, proinsulin or similar biomarkers • Limitations • Sufficient sample size • The massive number of statistical tests performed presents an unprecedented potential for thepositiveresults • Search the entire genome-->not worth the expenditure
Advantage of ExomeSequecing • Whole genome sequencing • Redundant raw data(6 Gb in each human diploid genome ) • Exomesequecing(targeted exome capture) • Exons are short and 180,000 exons constitute 1% of the human genome • The goal is to identify the functional variation that is responsible for both mendelian and common diseases
Significance • Exomesequencing can be used to identify causal variants of rare disorders • The first reported study that used exome sequencing as an approach to identify an unknown causal gene for a rare mendeliandisorder
The Shendure Lab • Next-generation human genetics • A multiplex approach to genome sequencing • Targeted sequence enrichment • Protocols relying on molecular inversion probe • Hybrid capture • Novel analytical strategies to identify the genetic basis of Mendelian disorders by exomesequecing • Autosomal recessive disorders such as Miller syndrome • Autosomal dominant disorders such as Kabuki syndrome
Hapmapproject • Focuse on common SNPs(at least 1% of the population) • Samples: 4 populations • (30*3 YRI, 30*3 CEU, 45 JPT, 45 CHB) • Data: • SNP frequencies, genotypes
Target enrichment Methods a. PCR-based approach b. Molecular inversion probe(MIP)-based approach c. Hybrid capture-based approach Mamanova et al. Nat Method 7(2):112-118
Mamanova et al. Nat Method 7(2):112-118
Figure. ①Probe list of array2 ② Probe list of array1 ③Exome on 1-22, X and Y chromosomes
Figure. Table of Splice Site Variants of 8 HapMap individuals
Figure. Table of coverage of 8 HapMap individuals and 4 FSS individual
Figure. ①Probe list of array2 ② Probe list of array1 ③Exome on 1-22, X and Y chromosomes
YRI: Nigeria - Yoruba people of Ibadan CHB: China - Beijing JPT: Japan - Tokyo CEU: Centre d'Etude du Polymorphisme Humain (CEPH) Eur: European–American ancestry
Considerations Causal genes may be shared by case group. Control group may not contain that mutation. Common mutation may not be causal. Causal mutation should cause animo acid change.
Further application Typical single gene disorder. Disorder caused by single but not uniform gene. Multiple gene disorder. Complex disease. Cancer.






















