Download
1 / 1
30 likes | 124 Views
Temperature effects through the chemical potential of oxygen. This study was supported by ERAF project Nr. 2010/0272/2DP/2.1.1.1.0/10/APIA/VIAA/088. Calculations of Electronic Structure of Defective ZnO: the impact of Symmetry and Phonons.

E N D
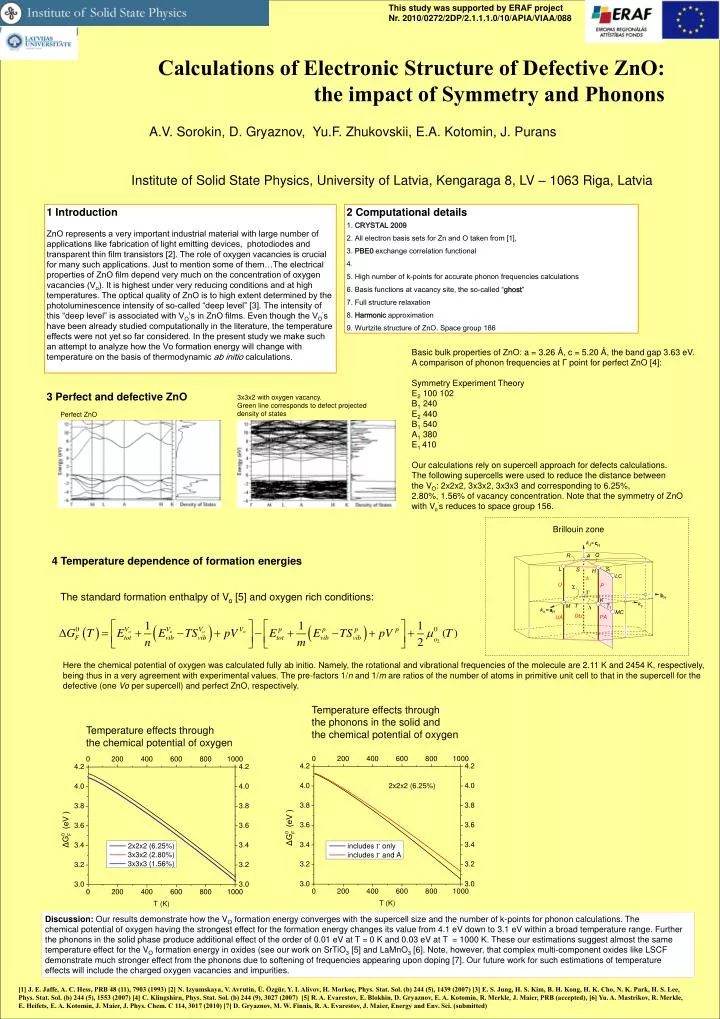
Temperature effects through the chemical potential of oxygen This study was supported by ERAF project Nr. 2010/0272/2DP/2.1.1.1.0/10/APIA/VIAA/088 Calculations of Electronic Structure of Defective ZnO: the impact of Symmetry and Phonons • V. Sorokin, D. Gryaznov, Yu.F. Zhukovskii, E.A. Kotomin, J. Purans Institute of Solid State Physics, University of Latvia, Kengaraga 8, LV – 1063 Riga, Latvia 1 Introduction ZnO represents a very important industrial material with large number of applications like fabrication of light emitting devices, photodiodes and transparent thin film transistors [2]. The role of oxygen vacancies is crucial for many such applications. Just to mention some of them…The electrical properties of ZnO film depend very much on the concentration of oxygen vacancies (Vo). It is highest under very reducing conditions and at high temperatures. The optical quality of ZnO is to high extent determined by the photoluminescence intensity of so-called “deep level” [3]. The intensity of this “deep level” is associated with VO’s in ZnO films. Even though the VO’s have been already studied computationally in the literature, the temperature effects were not yet so far considered. In the present study we make such an attempt to analyze how the Vo formation energy will change with temperature on the basis of thermodynamic ab initio calculations. 2 Computational details 1. CRYSTAL 2009 2. All electron basis sets for Zn and O taken from [1], 3. PBE0 exchange correlation functional 4. 5. High number of k-points for accurate phonon frequencies calculations 6. Basis functions at vacancy site, the so-called “ghost” 7. Full structure relaxation 8. Harmonic approximation 9. Wurtzite structure of ZnO. Space group 186 Basic bulk properties of ZnO: a = 3.26 Å, c = 5.20 Å, the band gap 3.63 eV. A comparison of phonon frequencies at Γ point for perfect ZnO [4]: Symmetry Experiment Theory E2 100 102 B1 240 E2 440 B1 540 A1 380 E1 410 Our calculations rely on supercell approach for defects calculations. The following supercells were used to reduce the distance between the VO: 2x2x2, 3x3x2, 3x3x3 and corresponding to 6.25%, 2.80%, 1.56% of vacancy concentration. Note that the symmetry of ZnO with Vo’s reduces to space group 156. 3 Perfect and defective ZnO 3x3x2 with oxygen vacancy. Green line corresponds to defect projected density of states Perfect ZnO Brillouin zone 4 Temperature dependence of formation energies The standard formation enthalpy of Vo [5] and oxygen rich conditions: Here the chemical potential of oxygen was calculated fully ab initio. Namely, the rotational and vibrational frequencies of the molecule are 2.11 K and 2454 K, respectively, being thus in a very agreement with experimental values. The pre-factors 1/n and 1/m are ratios of the number of atoms in primitive unit cell to that in the supercell for the defective (one Vo per supercell) and perfect ZnO, respectively. Temperature effects through the phonons in the solid and the chemical potential of oxygen Discussion: Our results demonstrate how the VO formation energy converges with the supercell size and the number of k-points for phonon calculations. The chemical potential of oxygen having the strongest effect for the formation energy changes its value from 4.1 eV down to 3.1 eV within a broad temperature range. Further the phonons in the solid phase produce additional effect of the order of 0.01 eV at T = 0 K and 0.03 eV at T = 1000 K. These our estimations suggest almost the same temperature effect for the VO formation energy in oxides (see our work on SrTiO3 [5] and LaMnO3 [6]. Note, however, that complex multi-component oxides like LSCF demonstrate much stronger effect from the phonons due to softening of frequencies appearing upon doping [7]. Our future work for such estimations of temperature effects will include the charged oxygen vacancies and impurities. [1] J. E. Jaffe, A. C. Hess, PRB 48 (11), 7903 (1993) [2] N. Izyumskaya, V. Avrutin, Ü. Özgür, Y. I. Alivov, H. Morkoç, Phys. Stat. Sol. (b) 244 (5), 1439 (2007) [3] E. S. Jung, H. S. Kim, B. H. Kong, H. K. Cho, N. K. Park, H. S. Lee, Phys. Stat. Sol. (b) 244 (5), 1553 (2007) [4] C. Klingshirn, Phys. Stat. Sol. (b) 244 (9), 3027 (2007) [5] R. A. Evarestov, E. Blokhin, D. Gryaznov, E. A. Kotomin, R. Merkle, J. Maier, PRB (accepted), [6] Yu. A. Mastrikov, R. Merkle, E. Heifets, E. A. Kotomin, J. Maier, J. Phys. Chem. C 114, 3017 (2010) [7] D. Gryaznov, M. W. Finnis, R. A. Evarestov, J. Maier, Energy and Env. Sci. (submitted)