Download

1 / 47
470 likes | 482 Views
Exam 2 Review: Neurodegenerative Diseases. Biology of Mental Disorders. Huntington’s Disease Lecture 1: Clinical Presentation. Trinucleotide repeat disease Autosomal dominant Genotype=Phenotype

E N D
Exam 2 Review: Neurodegenerative Diseases Biology of Mental Disorders
Huntington’s Disease Lecture 1: Clinical Presentation • Trinucleotide repeat disease • Autosomal dominant • Genotype=Phenotype • Affects men and women across all races/ethnic groups, but most common in people of European descent • Motor impairments: Chorea(“dance-like” movements) uncontrolled, jerky movement of arms, legs, head, upper trunk • Cognitive impairments: impairments in reasoning skills, memory, concentration, judgment, organization ability • Mood changes: depression, anxiety, anger/irritability
Huntington’s Disease Lecture 1: general genetic features • Mutation in huntingtin gene (HTT) on chromosome 4 • Huntingtin has polyglutamine “polyQ” triplet “CAG” codon repeat sequence • Repeat creates “sticky”, large intracellular aggregates • Expansions due to DNA polymerase “slipping” events • Anticipation • Juvenile HD: >60 repeats • Adult HD: >36 repeats
Huntington’s Disease Lecture 1: Gross Anatomy • Progressive neurodegeneration • Huge ventricles, severe striatal/basal ganglia atrophy, general gray matter thinning • Loss of GABA-ergic, medium spiny neurons (MSN’s) in striatum • May target striatum due to glutamate overaccumulation, leading to excitotoxicity • increased glu release increased Ca2+increased neuron death
Direct and Indirect Pathways: Huntington’s HUNTINGTON’S NORMAL FUNCTIONING Cortex Cortex Putamen D2 D1 Thalamus Thalamus Putamen D2 D1 Direct pathway Direct pathway SNc SNc Indirect pathway Indirect pathway GPe GPe Gpi/SNr Gpi/SNr Red = Inhibitory Green = Excitatory STN STN
Huntington’s Disease Lecture 2: Gain of Function • Gain of function features of Htt gene: expanded polyQ enables protein to do something it wouldn’t normally do • Higher levels of mHTT protein correlate with disease phenotypes • Expression of polyQ HTT fragments is toxic • Gene knockout in mice results in different anatomical and early life abnormalities that aren’t seen in people with the expanded allele Mice with different expression levels of same 120Q expansion: Higher protein expression less time on rotarod
Huntington’s Disease Lecture 2: Caspase 6 • Expression of cleaved caspase6fragments can result in disease • Cleavage fragments decreased latency to fall • Caspase-6 activity increased with mHTT expression in mice • Mutated caspase-6 cleavage site (C6R) is less harmful (looks like WT) Caspase 6 fragment with 86 Q repeat (C63) mice Caspase 6 fragment with 23 Q repeat (A2) mice Control mice
Huntington’s Disease Lecture 2: Loss of Normal Function • Loss of function features of Htt gene: polyQ prevents protein from doing what it normally does • Htt KO is lethal • Heterozygous mice are hyperactive & have cognitive deficits • Homozygosity for expansion is indistinguishable from heterozygosity in humans and not lethal in mice (expansion affects adult functions not developmental functions)
Huntington’s Disease Lecture 2: How Expanded Htt alters normal functions • Apoptosis: • Expanded Htt prevents cells from undergoing apoptosis when exposed to oxidant • Excitotoxicity: • Expanded Htt Inability to restore resting membrane potential • Expanded Htt Reduced ATP/ADP ratio • Expanded Htt Increased NMDA currents • Expanded Htt cells more susceptible to glutamate excitotoxicity • Altered nuclear function: • Nuclear export sequence aggregates in nucleus (rather than cytoplasm) • Reduction in BDNF • Neuronal transport: • Normal Htt important for shuttling mRNA and proteins & transcriptional regulation (NRSF/REST) • Mitochondrial health: • Expanded Htt Impaired resting mitochondrial membrane potential • Inhibit cytochrome c release with CSA (mitochondria transition core blocker) prevents excitotoxicity
Huntington’s Disease Lecture 2: histone deacetylase (HDAC) inhibitors • Rationale: • Huntingtin protein interacts with histone acetyltransferase (HAT) proteins, like CREB-binding protein (CBP) • CBP is found in nuclear inclusions with mutant htt • Mutant htt • Leads to less histone acetylation, recruitment of HAT complexes to DNA, lower gene expression • Inhibiting deacetylasesmay improve function
Huntington’s Disease Lecture 2: Disease Models • Drosophila eye model • Examine differences in histone acetylation • Large libraries of small molecule screens in C. elegans • Mouse models WT Poly-Q suppressor=hsp70 Mechanistic Therapeutic
Alzheimer’s Disease Lecture 1: Clinical Presentation • Impaired: Complex attention, executive function, learning and memory, language, motor perception and social cognition • Age-dependent (exponential increase after 6th decade of life) • African American and Hispanic individuals at higher risk AAMI MCI Cognitive Performance AD Age
Alzheimer’s Disease Lecture 1: Pathology Tau tangles Amyloid plaque • Pathology most likely begins before diagnosis • Extracellular amyloid plaques & tau tangles • Hippocampal/cortical degeneration (apoptosis, excitotoxicity, mitochondrial dysfunction, calcium dynamics, inflammation, etc…) • AD targets large pyramidal neurons (ex: CA1 and CA3 in hippocampus) • Cerebellum largely spared Asymptomatic Symptomatic Unresponsive substrate Responsive substrate?
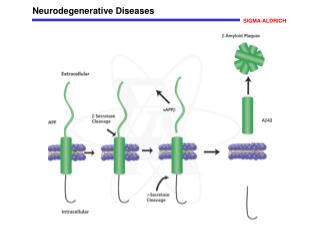
Alzheimer’s Disease Lecture 1: Pathology (continued) • Abeta accumulation: • Reduced clearance • Reduced degradation (Ubiquitin proteasome pathway) • Inflammation • Mutations in APP, PS1, PS2 • APOE4allele (APOE2 is protective) • α-,β-, and γ-secretase: • α=good, β=bad, γ=neutral • Most issues with Abeta42, issues with Abeta40; go on to form oligomers and plaques
Alzheimer’s Disease Lecture 2: Cholinergic Hypothesis • Basal forebrain cholinergic (CBF) cell death correlates with disease severity, pathology, synapse loss • CBF neurons depend on NGF for survival/maintenance • Hypothesis: inability to obtain or use NGF underlies demise of CBF neurons • AD may prevent retrograde transport of NGF in cholinergic neurons (decreased NGF receptors & ACh+ neurons with age) • Reduced NGF signaling may lead to Abeta accumulation
Alzheimer’s Disease Lecture 2: Amyloid hypothesis • Aβ: • Primary component of senile plaques, disrupt neuronal function neuronal death • May shift caspase-3 activity with age from “plasticity path”(protective factor) to a “cell death path” (pathogenic factor) • May spread in a “prion-like” manner • Gain of function
Alzheimer’s Disease Lecture 2: Disease Models • Amyloid precursor protein (APP) important for: • Axonal transport • Cell adhesion • Cell growth • Synaptic plasticity • Mutations in APP, PS1, and PS2are hallmarks offamilial AD • Have Abeta plaques, loss of synapses, spatial memory impairments, gliosis • Combining APP + PS1 earlier onset • APP KO mice indistinguishable from WT (implies plaques, etc… = gain of function) • Triplicate mouse model (3xTg) has beta secretase, APP, and familial mutation in tau gene • LTP deficits (weaker LTP with age)
Alzheimer’s Disease Lecture 2: BACE and HDAC inhibitors • BACE (beta-site APP-Cleaving Enzyme) inhibitor • beta secretase inhibitor • Reduces Abeta production • BACE KO mice have decreased pathology • BACE KO rescues memory defect in mice • HDAC (histone deacetylase) inhibitor • Preserves learning and memory performance in mice • Restores “lost” memory in mice
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD • Synaptic NMDAR: • Activated by low-frequency stimulation by glutamate released from vesicles • Activate CREB, stimulate BDNF, neuroprotective • More a-secretase, less abeta • Extrasynaptic NMDAR: • Activated by spillover glutamate at dendrite shaft, cell body • Decrease CREB, BDNF, dysregulate mitochondria, glutamate toxicity • Less a-secretase, increased abeta, & increased BACE • Importance of where glutamate enters
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD • Memantine/Nitromemantine: targets extrasynaptic NMDAR’s • Decrease abeta levels • Decrease phospho-tau levels • Blocks ROS & mitochondrial Ca2+ overload • Prevents CREB shut off • Questions: • Does A-beta stimulate glutamate release & where? Is it synaptic or extrasynaptic? • What happens in post-synaptic neurons after glutamate release? • Does A-beta affect synaptic plasticity? • What happens in spines after a-beta activation of eNMDAR?
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD • Does A-beta stimulate glutamate release and where? Is it synaptic or extrasynaptic? • It engages α-7 nicotinic acetylcholine receptors to release astrocytic glutamate extrasynaptic NMDA receptors on neurons • What happens in post-synaptic neurons after glutamate release? • Increases in calcium and nitric oxide • Chronic A-beta exposure in vivo shows increased glu baseline current • Does A-beta affect synaptic plasticity? • In hippocampus, eNMDAR activity is followed by reduction in evoked and miniature excitatory postsynaptic currents (mEPSC’s) • What happens in spines after a-beta activation of eNMDAR • Greater presence of phosphorylated tau • In vivo synapse loss • Memantine and nitromemantine rescue functions of above features
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD
Alzheimer’s Disease Journal Club: REST overexpression • REST: • Chromatin modifying silencing factor • Represses neurogenesis in embryonic development • Expressed in NSC’s; diminished when neurons mature • Overexpression blocks migration and neuronal differentiation • Goals: • Create conditional REST overexpression knock-in mouse • Determine role of REST • Determine relationship between REST and DRD2 gene • Measure locomotion
Alzheimer’s Disease Journal Club: REST overexpression • Made knock-in mouse • Cre-loxp model • Gene that REST regulates • See genome shift (from genes modifying postsynaptic membrane potential to gene modifying regulation of immune response) • REST seems to target DRD2 • Strong binding of DRD2 in striatal neurons • Relationship between REST and DRD2 • REST represses DRD2 • Locomotor findings • Increased immobility, decreased speed, and decreased total distance traveled by REST overexpression + DRD2 mice • Locomotion findings not due to neuronal or glial cell death/morphology changes, or apoptosis—confirms that locomotor findings are due to relationship between REST and DRD2
Parkinson’s Disease Lecture 1: Clinical Presentation • Chronic, progressive neurodegenerative disease that primarily affects movement • Age-dependent (60 yo; young-onset = <40 yo) • More frequent in men • Highly variable severity • Not immediately fatal; die from complications • Cardinal Motor Features: • (Resting) Tremor • Rigidity • Akinesia/bradykinesia • Postural Instability • Non-motor symptoms: • Anosmia • Depression/anxiety • Cognitive problems (focused attention, planning, language/memory) • Digestive issues • Fatigue • Sleep disorders
Parkinson’s Disease Lecture 1: Pathology • Progressive loss of dopaminergic neurons in the substantia nigra (SN) & nigrostriatal tract • α-synuclein (and ubiquitin +) deposits called Lewy Bodies aggregate as a result of dysfunction of ubiquitin proteasome system and lysosomal autophagy Lewy Body
Direct and Indirect Pathways: Parkinson’s PARKINSON’S NORMAL FUNCTIONING Cortex Cortex Putamen D2 D1 Putamen D2 D1 Thalamus Thalamus X Direct pathway Direct pathway SNc SNc Indirect pathway Indirect pathway GPe GPe Gpi/SNr Gpi/SNr Red = Inhibitory Green = Excitatory STN STN
Parkinson’s Disease Lecture 1: Genetic Causes • Mutation on chromosome 4 at SCNA gene(codes for α-synuclein) • Results from point mutations, triplication • α-synuclein present at presynaptic vesicles and promotes vesicle docking and fusion • Can form lipid-based aggregates • Excess α-synuclein can be released extracellularly—too much α-synuclein can be endocytosed by neighboring cells, leading to propagation • LRRK2, also Parkin, DJ-1, PINK-1, and ATP13A2 • Mutations in GBA(glucosylceramidasebeta) gene (important for lysosome function and autophagy) promotes α-synuclein propagation
Parkinson’s Disease Lecture 1: Environmental Causes • Herbicides (paraquat) • Pesticides (rotenone, permethrin, organochlorines) • Rural environments • Consuming well water • Proximity to industrial plants/quarries • Metal exposure (high-dose manganese & chronic lead exposure)
Parkinson’s Disease Lecture 2: MPTP Models • Neurotoxin MPTP mimics pathophysiology of PD byselectively targeting DA neurons in SN; inhibiting complex 1 of mitochondria reduction in TH+ neurons • Chronic MPTP administration: • Lewy bodies (α-synuclein & ubiquitin deposits) • Gliosis • Behavioral deficits • Apomorphine (APO): dopamine receptor agonist that rescues loss of DA neurons due to MPTP
Parkinson’s Disease Lecture 2: Genetic Models • Point Mutations causing accelerated aggregation of α-synuclein • A53T (more potent) • A30P • Overexpression • α-synuclein triplication (SCNA X 3) • Gain of function • Point mutation/overexpression models (rodentand fly) show motor deficiencies
Parkinson’s Disease Lecture 2: α-synuclein neurobiology • α-synuclein KO protective against MPTP • Effects of α-synuclein aggregates: • Disrupts proteosome activity; increases susceptibility to protein degradation • Disrupts mitochondrial respiration (complex I dysfunction increased ROS cytochrome c release apoptosis) • Binds to and depletes chaperones • Exacerbates dopamine-induced cell death • Altered vesicular recycling
Parkinson’s Disease Lecture 2: PD genes • PINK1 andParkinwork together to induce mitophagy, mutations here cause issues • LRRK2: important for synaptic vesicle formation; KO’s have fewer and smaller vesicle size with altered vesicular recycling
Parkinson’s Disease Lecture 2: Current Treatments and neuroprotective features • Most common 1st line of treatment: L-DOPA • Combined with carbidopa to prevent metabolism outside of brain and increase efficiency/availability of L-DOPA crossing BBB • Stem cell transplants • Deep brain stimulation • Thin wire electrode in STN or Gpi that delivers electrical pulses • Blocks over activation of Gpi/SNr • Neuroprotective features: • Nicotine • BDNF treatment • Exercise
Parkinson’s Disease Journal Club: Gut microbiota • 80% of PD patients have GI symptoms • Microbiome: bacteria that live in gut/intestines • Diversity is key • Question: Do peripheral symptoms in PD connect to findings in brain? • Vagus nerve • How does gut microbiome influence brain function in PD mouse models? • Goals: • Do gut microbiomes effect locomotor behavior? • What is the effect of gut microbiome on α-synuclein pathology? • Are there signs of CNS inflammation caused by microbiome? • Are effects of gut microbiome something determined in early life or is it an active, lifelong communication?
Parkinson’s Disease Journal Club: Gut microbiota • Do gut microbiomes effect locomotor behavior? • alpha synuclein overexpression (ASO) without gut bacteria (GF): • locomotion and GI movements like wild-type mice • Short-chain fatty acids (SCFA’s) seem to “mediate” PD gut-brain signaling • What is the effect of gut microbiome on α-synuclein pathology? • GF-ASO mice have limited/no α-synuclein deposits; same α-synuclein production as SPF-ASO mice, but different handling • Signs of inflammation caused by microbiome? • GF-ASO mice microglia look like wt (more branches, more branching, more “space-filling”) • Are effects of gut microbiome something determined in early life or is it an active, lifelong communication? • Active, lifelong communication • Human fecal transplants: mouse resembled phenotype of human donor