Download

1 / 69
720 likes | 898 Views
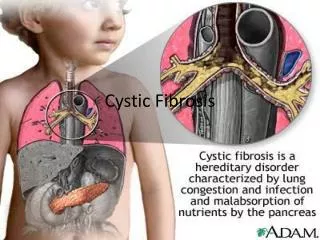
Cystic Fibrosis. Dr. Daksh Jhim First Year Junior resident. introduction. Cystic fibrosis (CF) is a monogenic disorder that presents as a multisystem disease. was first described as a unique disease entity in 1938. Most common lethal genetic disease in Causasians .

E N D
Cystic Fibrosis Dr. DakshJhim First Year Junior resident
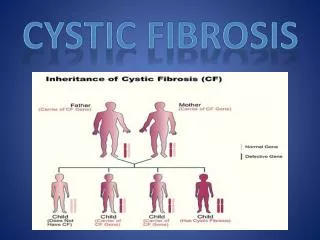
introduction • Cystic fibrosis (CF) is a monogenic disorder that presents as a multisystem disease. • was first described as a unique disease entity in 1938. • Most common lethal genetic disease in Causasians. • lethal autosomal recessive disease. • incidence: 1 in 2000-3000; predominantly Caucausian populations (carrier frequency 1 in 22-28). • “Woe to the child which when kissed on the forehead tastes salty. He is bewitched and soon will die” - old proverb.
introduction • Patients typically presented with intestinal obstruction or malnutrition and died from overwhelming pneumonia within the first year of life. • Over the last 40 years, the median survival with CF has increased dramatically from 6 years in 1955 to 36 years in 2005. • The improvement in CF outcomes has paralleled advances in antibiotic therapies, nutritional approaches, and the collection of clinical expertise into specialized treatment centers.
CF and genetics • Caused by mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. • Anion channel • ATP Binding Cassette family • Regulates other ion channels (ENaC) • Plays crucial roles in absorption and secretion • Found mainly in wet epithelia
CFTR • CFTR functions as a cAMP-activated ATP-gated anion channel. • increasing the conductance for certain anions (e.g. Cl–) to flow down their electrochemical gradient. • ATP-driven conformational changes in CFTR open and close a gate to allow transmembrane flow of anions down their electrochemical gradient. • found in the epithelial cells of many organs including the lung, liver, pancreas, digestive tract, reproductive tract, and skin. • Mainly found in exocrine glands. • Normally, the protein moves chloride and thiocyanate ions along the concentration gradient.
Sweat glands • Mucus • Digestive fluids
Sweat gland pathophsiology • CFTR important for Cl- absorption • Na+ and Cl- generally reabsorbed • Cl- buildup = negative charge on luminal side • Decreased Na+ reabsorption • NaCl formation and secretion in sweat • Used as diagnostic factor
mutations • The CFTR gene located on chromosome 7, on the long arm at position q31.2.. • The mutations in the CFTR gene fall into five major classes. • Classes I–III mutations are considered "severe," as indexed by pancreatic insufficiency and high sweat NaCl values. • Class IV and V mutations can be "mild," i.e., associated with pancreatic sufficiency and intermediate/normal sweat NaCl values.
mutations • Over 1900 mutations in the CFTR gene that can cause CF. • Most common is ΔF508.
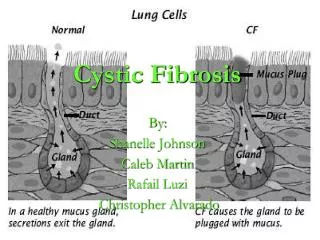
LUNG PATHOPHYSIOLOGY • CFTR plays a central role in the regulation of ion transport across airway epithelia. • In CF, dysregulation of airway surface liquid (ASL) volume occurs as a consequence of ion transport dysfunction • impairing mucociliary clearance and, therefore, lung defense. • Airway surfaces are coatedwith a thin layer of liquid, the ASL, which is composed of a • periciliary layer (PCL) • more viscous mucus layer.
LUNG PATHOPHYSIOLOGY • The mucus layer, which normally floats on top of the PCL, efficiently traps inhaled pathogens and particulates. • The mucus layer, which normally floats on top of the PCL, efficiently traps inhaled pathogens and particulates. • The underlying PCL layer, in turn, provides a low viscosity environment in which cilia can beat freely and thereby propel the mucus layer toward the mouth. • The PCL also acts as a lubricant layer that prevents adhesion of the mucus layer to cell surfaces. • proper regulation of ASL volume and the hydration of its component layers are critical to the maintenance of mucus clearance.
LUNG PATHOPHYSIOLOGY • An adequate PCL height is necessary to allow ciliary beating. • Adequate hydration of the mucus layer is a key determinant of its viscoelastic properties and transportability.
LUNG PATHOPHYSIOLOGY • CFTR important for Cl- secretion • CFTR also mediate ENaC activity • Net decrease in Cl- secretion and increase in Na+ absorption • Osmoticallysurface mucous dehydrated • Buildup compresses cilia resulting no mucous movement • Infections.
Overall pathology • Sweat Glands :hypersecretion of salt • Lungs :mucous buildup + infection • Pancreas :fibrous cysts + blockage • GI Tract :blockage from thick feces • Liver : obstruction, possible cirrhosis • Male Reproduction : sterile
Lung :secondary pathogenesis • ASL dehydration produces progressive mucostasis. • Initiates a cascade of events that leads to clinically apparent CF lung disease. • Firstly, thickened mucus secretions eventually become adherent to airway surfaces with the loss of PCL volume, and begin to obstruct small airway lumens. • Mucus plugs and plaques not only provide a protected environment in which bacteria can escape mechanical and immune-mediated clearance, but also create a unique environment that drastically alters bacterial gene expression.
Lung :secondary pathogenesis • Paradoxically, the center of a mucus plug is in fact anaerobic (pO2 less than 2 Torr) due to a combination of an increased diffusion distance for O2 as well as increased oxygen consumption by CF epithelia (owing to heightened Na+ transport). • Within this environment, P. aeruginosa converts to an anaerobic mode of metabolism, increases alginate production, and ultimately establishes a biofilm structure. • Organisms growing within the biofilm possess increased resistance to secondary host defense mechanisms (e.g., neutrophils and soluble antimicrobials).
Failure of neutrophils to clear this infection, accompanied by the release of proteases and other harmful substances destroying lung tissue, ultimately leads to bronchiectasis.
Clinical aspects ASL dehydration and mucus plugging, also develops chronic airway inflammation without readily identifiable bacterial infection. • Current hypothesis to explain these findings include: • a low burden of typical bacteria avoids eradication in the dehydrated mucus environment of the CF lung and drives the inflammatory process in very early disease. • the presence of atypical, perhaps anaerobic, organisms are poorly identified with usual culture systems but dominate early disease and cause inflammation. • intermittent events, including viral infections and/or gastric aspiration drive the inflammatory process early in life. • Regardless, these observations point to the very early onset of lung disease, even in asymptomatic infants, and the need to develop and institute effective interventions in this population.
Clinical aspects • Over a relatively short period of time, the CF lung becomes chronically infected with typical pathogens. • In childhood, Haemophilusinfluenzae and Staphylococcus aureusare often identified first organisms to invade typically followed by the establishment of chronic Pseudomonas aeruginosainfection. • Pseudomonas may take many morphologic forms, but mucoid phenotype signifies chronicity and the inability to eradicate this organism, even with aggressive antibiotic regimens. • Other important pathogens that are encountered in CF include a variety of gram-negative bacteria, especially Stenotrophomonasmaltophilia, Achromobacterxylosoxidans, and the Burkholderiacepacia complex (Bcc).
Clinical aspects • Mycobacterial pathogens are also encountered in CF, including M. avium complex and M. abscessus. • MAC is the most prevalent mycobacterial pathogen in CF, but often does not cause discernible clinical disease, as opposed to the much more problematic infection with M. abscessus. • Viral infections, although probably not more frequent than in other populations, do appear to cause more morbidity and may be an important trigger of lung disease exacerbations. • Fungi, particularly aspergillus species, are common colonizers but may cause allergic bronchopulmonaryaspergillosis (ABPA).
The primary cause of morbidity and mortality in CF patients is bronchiectasis and obstructive lung disease. • Pulmonary disease is present in 98% of patients with CF by the time they reach adulthood. • A recurrent cough that becomes persistent is often the first manifestation. • Airway hyperreactivity and wheezing are common in children. • Pansinusitiswith opacificationof the paranasal sinuses is a universal finding in nearly 30% patients with CF.
Acute exCerbations • Exacerbations of CF lung disease are extremely important events in the life of a CF patient. • These periodic illnesses often remove the patient from their usual work or school activities, are associated with significant reductions in quality of life, exact a large financial toll in terms of health care costs, and are associated with reduced survival. • Exacerbations are typically acute to subacute events that are superimposed upon a previously stable clinical baseline.
Patients usually report increased cough, sputum, fatigue, and weight loss during these episodes. • Fever, leukocytosis, chest pain, and new infiltrates on chest radiographs are less consistent findings with exacerbations. • The inciting events that trigger an exacerbation have not been clearly defined, although acute respiratory viral infection may be one important cause in addition to inadequate use of preventative therapies.
Respiratory complications • Bronchiectasis: occurs as a result of destruction of lung tissue and erosion of the bronchial cartilage. • Atelectasis: Lobar and segmental atelectasis occurs in about 5% of patients. This complication is most prevalent in the first 5 years of life and thereafter has a diminishing frequency. The right lung is the site of atelectasis in the majority of patients. • Pneumothorax : is a more frequent complication .The overall incidence is about 1%/yr, and increases with age and disease severity, so that about 20% of CF adults will experience at least one pneumothorax.
Lung complications 4. Hemoptysis:is a common event in older CF patients. Minor to massive hemoptysis may occur .Massive hemoptysis is increasingly common in older patients, with an average annual incidence of 0.87 percent. 5. Allergic bronchopulmonaryaspergillosis (ABPA): develops in 1% to 10% of patients at some time in their lives. 6. Pleural effusions and empyema: are uncommon in patients with CF, but pleuritic symptoms and signs may accompany exacerbations of lung disease.
Staphylococcal and pseudomonalempyemas have been described, but respiratory tract infections usually spare the pleural space. 7. Respiratory failure: leads to death in greater than 90% of CF patients.
Radiological findings • The earliest radiographic change is usually hyperinflation of the lungs, reflecting obstruction of small airways. • The degree of hyperinflation generally increases with age. • As bronchitis progresses, peribronchial cuffing becomes increasingly prominent. • Mucous impaction in airways may be seen as branching finger-like shadows. • Evidence of bronchiectasis, such as enlarged ring shadows and cysts, is common by 5 to 10 years of age. • peripheral rounded densities are noted during acute exacerbations and may disappear with treatment, leaving residual cysts.
Radiological findings • Subpleuralblebs often become evident during the second decade of life and are most prominent along the mediastinalborder. • For reasons that remain unexplained, the right upper lobe usually displays the earliest and most severe changes. • With advancing disease, the pulmonary artery segments become more prominent. • A relatively small and vertical cardiac shadow is typical, but the heart enlarges with onset of corpulmonale. • Hilaradenopathy is rarely prominent. • Lobar or segmental atelectasis is uncommon but is most often seen in infants or small children.
Pulmonary function tests • Newborns with CF are thought to have normal lung function. • However, within weeks to months, many infants with CF show evidence of increased airway resistance, gas trapping, and diminished flow rates. • In general, patients progress from initial reductions in maximum midexpiratory flow rates to reductions in forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) and then to diminished vital capacity and total lung volumes. • The progression occurs from peripheral airway obstruction to more generalized obstruction and then to acquisition of a restrictive component.
Diagnostics • New born screening • Since October 2007, newborn screening for CF has been in place throughout the whole of the UK. • NHS of U.K. has made it mandatory for screening of newborns for cystic fibrosis. • Immunoreactivetrypsinogen (IRT) is measured on a dried blood spot obtained on the Guthrie card at day 6 of life. • Samples with abnormally raised IRT levels will undergo CFTR mutation screening as per the flow chart • An increased IRT must be followed by direct diagnostic testing (typically with the sweat chloride test).
diagnostics • Sweat testing(pilocarpineiontophoresis) • remains the “gold standard” diagnostic test because genetic screening only identifies a small number of the most common mutations. • The results of this test are abnormal in the large majority of patients with CF.(about in 98% of patients). • However, the test must be repeated at least twice, and an adequate sample containing at least 75 mg of sweat must be collected over a 30-min period. • Approximately 1% of patients with CF have normal sweat chloride test results.
False positive sweat test results • Adrenal insufficiency • Anorexia nervosa • Atopic dermatitis • Autonomic dysfunction • Celiac disease • Familial cholestasis • Fucosidosis • Glucose-6-phosphate dehydrogenase deficiency • Glycogen storage disease, type I • Hypogammaglobulinemia • Hypoparathyroidism • Hypothyroidism (untreated) • Klinefelter syndrome • Mucopolysaccharidosis, type I • Malnutrition • Nephrogenic diabetes insipidus • Nephrosis • Prostaglandin E1 infusion, long-term • Pseudohypoaldosteronism
Diagnostics 3. Molecular diagnosis: according to GUIDELINES, reasons for full genotyping include the following: • Any child diagnosed with CF: • facilitates screening for other family members. • allows prenatal diagnosis of future pregnancies. • Since the advent of the first mutation-specific therapy ivacaftor, and on-going clinical trials of other small molecule CFTR-modulators, all CF patients MUST be genotyped. Full gene sequencing should be carried out when there is diagnostic doubt (especially in ethnic minorities). • In newborn siblings of affected children, cord blood should be taken at the time of birth (arrange with mother in clinic, give form and blood bottle).
Generally older siblings will have a sweat test for diagnosis rather than genetic analysis. The latter would detect carriers, which is something that should be postponed until the sibling is old enough to decide whether they wish to know their carrier status (usually mid teens and older). • To aid confirmation of diagnosis in case of borderline sweat test. However, genotype analysis should not be used to guide prognosis in an individual child, except rarely (and very cautiously) in the case of mutations usually associated with pancreatic sufficiency (e.g. R117H).
Limitations of mutation analysis • Due to the large number of identified mutations, and the extreme rarity of many of these, it is only practical to screen for a few on a routine basis. • Clearly therefore failure to detect mutations does not exclude the diagnosis. The above is of particular importance in a child of non-Caucasian origin. • There is now a specific panel of mutations, which are common in the Asian community. • It is therefore CRITICALLY IMPORTANT that in every case the child’s ethnic origin is included on the request form so that the most likely mutations can be looked for.
Diagnostics • Potential Diff. across the respiratory epithelium • This is a research tool to aid in the diagnosis of CF. • Difficult in small children as requires cooperation, but may be useful in older indeterminate cases (over 8-10 years). • The abnormal transport of chloride leads to a very negative PD across the nasal epithelium. • Nasal perfusion with amiloridehydrochloride and with chloride-free solutions leads to characteristic changes in the PD.
Stool elastase: • may be supportive of the diagnosis. • low in CF with pancreatic insufficiency (usually <15 mcg/g). • Normal : > 200 mcg/g stool • Mild/moderate pancreatic insufficiency :100-200 mcg/g stool • Severe pancreatic insufficiency :< 100 mcg/g stool
Diagnostic criteria according to ACCP • Elevated sweat chloride level ≥ 60 mmol/L on two occasions or • Identification of mutations known to cause CF in both CFTR genes or • In vivo demonstration of characteristic abnormalities in ion transport across the nasal epithelium plus • One or more phenotypical features of CF • Sinopulmonary disease • Characteristic GI or nutritional disorders • Obstructive azoospermia • Salt loss syndrome or • Sibling with CF or • Positive NBS result
TREATMENT • The primary objectives of CF treatment are to control infection, promote mucus clearance, and improve nutrition. • Experience has repeatedly demonstrated that attention to preventive aspects of lung care and psychosocial factors is important.
Physical Measures to Promote Airway Clearance • The use of postural drainage with chest percussion to clear mucus is based on the concept that cough clears mucus from large airways but chest vibrations are necessary to move secretions from the small airways where expiratory flow rates are low. • When patients were receiving chest physical therapy on a regular basis, the only immediate effect documented was an increase in peak expiratory flow rate 30 minutes after therapy. • However, after 3 weeks without chest physical therapy, both FVC and flow rates were significantly reduced. • Theoretically, chest physical therapy might prevent or delay inspissation of mucus in small airways.
Inhaled Hypertonic Saline for Airway Clearance • Inhaled hypertonic saline (HS) osmotically draws water onto airway surfaces. • Indeed, because of the limitation with respect to Cl- permeability of CF airway epithelia, inhaled salt may remain on CF airway surfaces longer, and hence be osmotically more active, than on normal airway surfaces. • Recent data suggest that the rehydrating effect of inhaled HS (7%, four times daily) produces short term (2-wk) effects that restore mucus clearance and improve lung function and quality of life.