Download
1 / 1
10 likes | 124 Views
Conditional Probability. Pairs of aminoacids (boundaries). Frequency. A C D E F G H I K L M N P Q R S T V W Y Aminoacids. Conditional Probability. Pairs of aminoacids (boundaries). Distance (Angstron). 1 10 20 30 40 Deleted size (aminoacids).

E N D
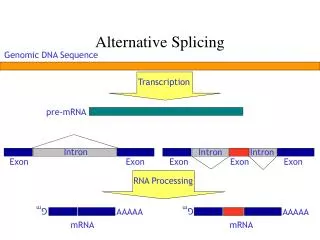
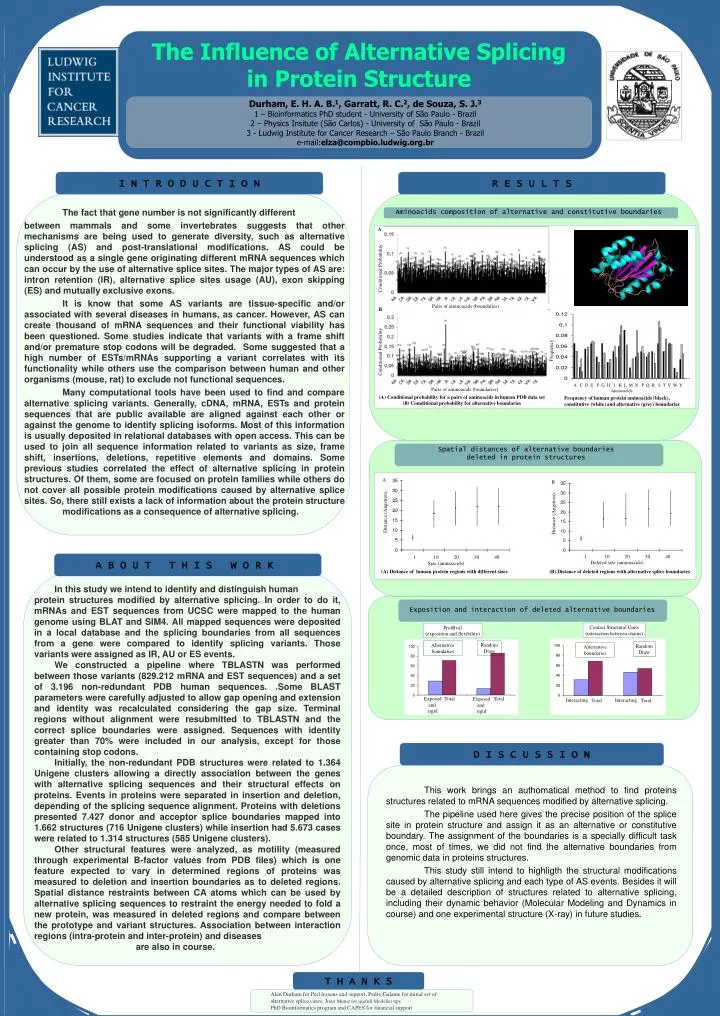
Conditional Probability Pairs of aminoacids (boundaries) Frequency A C D E F G H I K L M N P Q R S T V W Y Aminoacids Conditional Probability Pairs of aminoacids (boundaries) Distance (Angstron) 1 10 20 30 40 Deleted size (aminoacids) The Influence of Alternative Splicing in Protein Structure I N T R O D U C T I O N R E S U L T S The fact that gene number is not significantly different between mammals and some invertebrates suggests that other mechanisms are being used to generate diversity, such as alternative splicing (AS) and post-translational modifications. AS could be understood as a single gene originating different mRNA sequences which can occur by the use of alternative splice sites. The major types of AS are: intron retention (IR), alternative splice sites usage (AU), exon skipping (ES) and mutually exclusive exons. It is know that some AS variants are tissue-specific and/or associated with several diseases in humans, as cancer. However, AS can create thousand of mRNA sequences and their functional viability has been questioned. Some studies indicate that variants with a frame shift and/or premature stop codons will be degraded. Some suggested that a high number of ESTs/mRNAs supporting a variant correlates with its functionality while others use the comparison between human and other organisms (mouse, rat) to exclude not functional sequences. Many computational tools have been used to find and compare alternative splicing variants. Generally, cDNA, mRNA, ESTs and protein sequences that are public available are aligned against each other or against the genome to identify splicing isoforms. Most of this information is usually deposited in relational databases with open access. This can be used to join all sequence information related to variants as size, frame shift, insertions, deletions, repetitive elements and domains. Some previous studies correlated the effect of alternative splicing in protein structures. Of them, some are focused on protein families while others do not cover all possible protein modifications caused by alternative splice sites. So, there still exists a lack of information about the protein structure modifications as a consequence of alternative splicing. Aminoacids composition of alternative and constitutive boundaries A B (A) Conditional probability for a pairs of aminoacids in human PDB data set (B) Comditional probability for alternative boundaries Frequency of human protein aminoacids (black), constitutive (white) and alternative (grey) boundaries Spatial distances of alternative boundaries deleted in protein structures A B Distance (Angstron) Durham, E. H. A. B.1, Garratt, R. C.2, de Souza, S. J.3 1 – Bioinformatics PhD student - University of São Paulo - Brazil 2 – Physics Insitute (São Carlos) - University of São Paulo - Brazil 3 - Ludwig Institute for Cancer Research – São Paulo Branch - Brazil e-mail:elza@compbio.ludwig.org.br 1 10 20 30 40 Size (aminoacids) A B O U T T H I S W O R K (A) Distance of human protein regions with different sizes (B) Distance of deleted regions with alternative splice boundaries In this study we intend to identify and distinguish human protein structures modified by alternative splicing. In order to do it, mRNAs and EST sequences from UCSC were mapped to the human genome using BLAT and SIM4. All mapped sequences were deposited in a local database and the splicing boundaries from all sequences from a gene were compared to identify splicing variants. Those variants were assigned as IR, AU or ES events. We constructed a pipeline where TBLASTN was performed between those variants (829.212 mRNA and EST sequences) and a set of 3.196 non-redundant PDB human sequences. .Some BLAST parameters were carefully adjusted to allow gap opening and extension and identity was recalculated considering the gap size. Terminal regions without alignment were resubmitted to TBLASTN and the correct splice boundaries were assigned. Sequences with identity greater than 70% were included in our analysis, except for those containing stop codons. Initially, the non-redundant PDB structures were related to 1.364 Unigene clusters allowing a directly association between the genes with alternative splicing sequences and their structural effects on proteins. Events in proteins were separated in insertion and deletion, depending of the splicing sequence alignment. Proteins with deletions presented 7.427 donor and acceptor splice boundaries mapped into 1.662 structures (716 Unigene clusters) while insertion had 5.673 cases were related to 1.314 structures (585 Unigene clusters). Other structural features were analyzed, as motility (measured through experimental B-factor values from PDB files) which is one feature expected to vary in determined regions of proteins was measured to deletion and insertion boundaries as to deleted regions. Spatial distance restraints between CA atoms which can be used by alternative splicing sequences to restraint the energy needed to fold a new protein, was measured in deleted regions and compare between the prototype and variant structures. Association between interaction regions (intra-protein and inter-protein) and diseases are also in course. Exposition and interaction of deleted alternative boundaries Contact Structural Units (interaction between chains) ProfBval (exposition and flexibility) Random Draw Alternative boundaries RandomDraw Alternative boundaries Exposed and rigid Total Total Exposed and rigid Interacting Interacting Total Total D I S C U S S I O N This work brings an authomatical method to find proteins structures related to mRNA sequences modified by alternative splicing. The pipeline used here gives the precise position of the splice site in protein structure and assign it as an alternative or constitutive boundary. The assignment of the boundaries is a specially difficult task once, most of times, we did not find the alternative boundaries from genomic data in proteins structures. This study still intend to highligth the structural modifications caused by alternative splicing and each type of AS events. Besides it will be a detailed description of structures related to alternative splicing, including their dynamic behavior (Molecular Modeling and Dynamics in course) and one experimental structure (X-ray) in future studies. T H A N K S Alan Durham for Perl lessons and support, Pedro Galante for initial set of alternative splice cases, Joao Muniz for usefull Modeller tips PhD Bioinformatics program and CAPES for financial support