Download
1 / 1
10 likes | 98 Views
Overview. Methods. Results. We present GlyPID 2.0, a software tool for accurate characterization of protein glycosylation through a combination of the following techniques Accurate precursor ion mass calculated using THRASH–based deisotoping methods [2, 3]

E N D
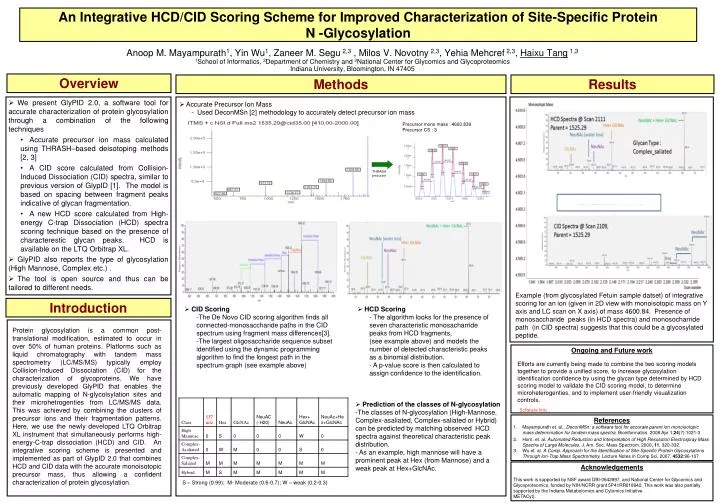
Overview Methods Results • We present GlyPID 2.0, a software tool for accurate characterization of protein glycosylation through a combination of the following techniques • Accurate precursor ion mass calculated using THRASH–based deisotoping methods [2, 3] • A CID score calculated from Collision-Induced Dissociation (CID) spectra, similar to previous version of GlypID [1]. The model is based on spacing between fragment peaks indicative of glycan fragmentation. • A new HCD score calculated from High-energy C-trap Dissociation (HCD) spectra scoring technique based on the presence of characterestic glycan peaks. HCD is available on the LTQ Orbitrap XL. • GlyPID also reports the type of glycosylation (High Mannose, Complex etc.) . • The tool is open source and thus can be tailored to different needs. • Accurate Precursor Ion Mass • - Used DeconMSn [2] methodology to accurately detect precursor ion mass Precursor mono mass : 4600.839 Precursor CS : 3 An Integrative HCD/CID Scoring Scheme for Improved Characterization of Site-Specific ProteinN -Glycosylation THRASH precursor Anoop M. Mayampurath1, Yin Wu1, Zaneer M. Segu 2,3 , Milos V. Novotny 2,3, Yehia Mehcref 2,3, Haixu Tang 1,3 1School of Informatics, 2Department of Chemistry and 3National Center for Glycomics and Glycoproteomics Indiana University, Bloomington, IN 47405 Example (from glycosylated Fetuin sample datset) of integrative scoring for an ion (given in 2D view with monoisotopic mass on Y axis and LC scan on X axis) of mass 4600.84. Presence of monosaccharide peaks (in HCD spectra) and monosocharride path (in CID spectra) suggests that this could be a glycosylated peptide. Introduction • HCD Scoring • The algorithm looks for the presence of seven characteristic monosacharride peaks from HCD fragments, • (see example above) and models the number of detected characteristic peaks as a binomial distribution. • A p-value score is then calculated to assign confidence to the identification. • CID Scoring • The De Novo CID scoring algorithm finds all connected-monosaccharide paths in the CID spectrum using fragment mass differences[3]. • The largest oligosaccharide sequence subset identified using the dynamic programming algorithm to find the longest path in the spectrum graph (see example above) Protein glycosylation is a common post-translational modification, estimated to occur in over 50% of human proteins. Platforms such as liquid chromatography with tandem mass spectrometry (LC/MS/MS) typically employ Collision-Induced Dissociation (CID) for the characterization of glycoproteins. We have previously developed GlyPID that enables the automatic mapping of N-glycolsylation sites and their microhetrogenties from LC/MS/MS data. This was achieved by combining the clusters of precursor ions and their fragmentation patterns. Here, we use the newly developed LTQ Orbitrap XL instrument that simultaneously performs high-energy-C-trap dissociation (HCD) and CID. An integrative scoring scheme is presented and implemented as part of GlypID 2.0 that combines HCD and CID data with the accurate monoisotopic precursor mass, thus allowing a confident characterization of protein glycosylation. Ongoing and Future work Efforts are currently being made to combine the two scoring models together to provide a unified score, to increase glycosylation identification confidence by using the glycan type determined by HCD scoring model to validate the CID scoring model, to determine microheterogenties, and to implement user-friendly visualization controls. • Prediction of the classes of N-glycosylation • The classes of N-glycosylation (High-Mannose, Complex-asaliated, Complex-saliated or Hybrid) can be predicted by matching observed HCD spectra against theoretical characteristic peak distribution. • As an example, high mannose will have a prominent peak at Hex (from Mannose) and a weak peak at Hex+GlcNAc. Software link: • References • Mayampurath et. al., DeconMSn: a software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics. 2008 Apr 1;24(7):1021-3 • Horn, et. al. Automated Reduction and Interpretation of High Resolution Electrospray Mass Spectra of Large Molecules. J. Am. Soc. Mass Spectrom. 2000, 11, 320-332. • Wu et. al. A Comp. Approach for the Identification of Site-Specific Protein Glycosylations Through Ion-Trap Mass Spectrometry, Lecture Notes in Comp Sci, 2007, 4532:96-107. Acknowledgements This work is supported by NSF award DBI-0642897, and National Center for Glycomics and Glycoproteomics, funded by NIH/NCRR grant 5P41RR018942. This work was also partially supported by the Indiana Metabolomics and Cytomics Initiative METACyt). S – Strong (0.99); M- Moderate (0.6-0.7); W – weak (0.2-0.3)