Download

1 / 32
400 likes | 811 Views
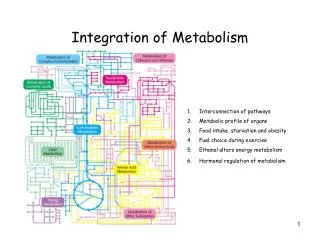
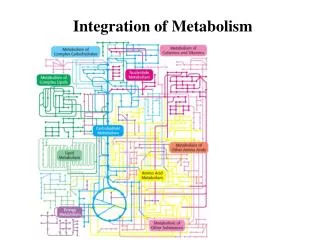
Integration of Metabolism. FUELS, METABOLITES AND DISORDERS FUELS URINE BILE/FECES METABOLITES METABOLITES Starch, Glucose NH 4 , + SO 4 2- , HPO 4 2- Cholesterol, Mannose Creatinine Bile Acids Sucrose (Fructose) Urea, Urate Bilirubin

E N D
FUELS, METABOLITES AND DISORDERS • FUELS URINE BILE/FECES • METABOLITES METABOLITES • Starch, Glucose NH4,+ SO42-, HPO42- Cholesterol, • Mannose Creatinine Bile Acids • Sucrose (Fructose) Urea, Urate Bilirubin • Amino Acids 17-Ketosteroids, -alanine Stercobilin • Glycerol -Aminoisobutyrate Lipid • Fat, Phospholipid (Steatorrhea) • Fatty Acids Ketone Bodies • Sorbitol Polyamines • Nucleic Acids(minor) Xanthurenic acid • Lactose(Galactose) Urobilinogen, Urobilin • Ketone Bodies 1-3
Metabolic Diseases and Metabolites Biotin deficiency: propionic acid, branched -ketoacids Amino Acids -->--> Propionic Acid ----> Succinyl CoA B12 deficiency: methylmalonate, homocysteine Hemolytic anemia and certain porphyrias: bilirubin PKU: Phe, phenyllactate, phenylacetate, phenylpyruvate Hypoxia: lactic acid Hyperammonemia: citrulline, argininosuccinate, etc. Biopterin or biopterin reductase deficiency: decrease in 5-hydroxy-indoleacetic, vanillyl mandelic acid, homovanillic acids 1-3
Metabolic Diseases and Metabolites (continued) Folic acid deficiency: formiminoglutamic acid (FIGLU) Diabetes mellitus: glucose (blood and urine), HbA1c, DKA Fructose intolerance: fructose or fructose 1-Pi *Cystinuria with basic A.A. renal transporter defect: cystine *Hartnup's disease: neutral aminoaciduria (Trp loss -> Niacin ) Homocystinuria: homocysteine Gaucher's disease: glucocerebrosides (lysosomal disorder)
1-3 Metabolic Disturbances and Fuel Intolerances Carbohydrate IntoleranceProtein Intolerance Diabetes mellitus Methylmalonyl aciduria Fructose Intolerance Maple Syrup urine disease Cushing's Disease (ACTH) Vitamin B12/Biotin deficiency Galactosemia Urea cycle deficiency Lactose Intolerance Phenylketonuria Lipid Intolerance LPL Def. (VLDL or Chylo ) MCAD
1- 4 Exercise Intolerance:Ketoacidosis/Ketonemia (Early vs. Prolonged) Decreased PFK-1 Diabetic ketoacidosis McArdles' disease Organic acidurias Carnitine, CAT1 deficiency Alcoholic ketoacidosis MCAD deficiency Glycogen storage disease
Glycogen Storage: Glucose 6-phosphatase (Type 1) - Von Gierke’s mild to severe hypoglycemia - only GNG tissues affected Lysosomal -glucosidase (Type 2) - Pompe’s no hypoglycemia --> fatal - all tissues affected Amylo-1,6-glucosidase (Type 3) - Cori’s mild hypoglycemia Glycogen Phosphorylase - McArdle’s no hypoglycemia - low lactate upon exercise Glycogen Synthase - hypoglycemia / hyperketonemia
1-4 Screening and Treatment of Metabolic Disease Disease Screening ProgramsMethods of Treatment Sickle Cell Disease * Dietary and Vitamin Therapy Tyrosinemia I Drug, hormone or metabolite Phenylketonuria * administration (allopurinol, Galactosemia * benzoic or phenylacetic acid, Homocystinuria insulin, carnitine, heme, etc.) Biotinidase Deficiency Enzyme replacement Maple syrup urine disease Genetic engineering (stem cell Adrenal hyperplasia replacement) Hypothyroidism (Cretinism) * Organ transplantation
Allosteric Regulation • Glycolysis: Hexokinase, Phosphofructokinase-1, • Pyruvate kinase (ATP, Ala) • Gluconeogenesis: Pyruvate carboxylase, • Fructose 1,6-bisphosphatase • Glycogenolysis: Glycogen phosphorylase kinase ( Ca2+) • Glycogen phosphorylase • (AMP, ATP, Glucose) • Glycogenesis: Glycogen Synthase (Glucose 6-P) • Fatty acid Synth: Acetyl CoA carboxylase • Beta Oxidation : CAT I (Malonyl CoA), Thiolase (Acetyl CoA) • -hydroxy fatty acyl CoA dehyd. (NADH) • Cholesterol Synth: HMGCoA reductase (Cholesterol) • Pyrimidine Synth: Carbamyl Pi Synthetase II (UTP) • Purine Synthesis : PRPP amidotransferase (Nucleotides) • Heme Synthesis:-Aminolevulinic acid synthase (Heme) • Ammoniagenesis: Glutamate Dehydrogenase (ADP/GDP) • Citric Acid Cycle Citrate Synthase, IC and KG Dehyd. • (NADH, ATP) • Urea Cycle Carbamyl Pi Synthetase I (N-Acetyl Glu)
1-4 Covalent Modification (Response of Activity to Phosphorylation, (Acute or Fine Control) Glycogen phosphorylase () Adipose triacylglycerol lipase () Glycogen synthase () Acetyl CoA carboxylase () Hepatic pyruvate kinase () HMG CoA reductase () Pyruvate dehydrogenase () Phosphorylase b kinase ()
1-5 Compartmentation Cytosolic: Glycolysis, pentose pathway, fatty acid / triacylglycerol synthesis, nucleotide synthesis, cholesterol biosynthesis Mitochondrial: TCA, electron transport, oxidation of fatty acids, ketone body formation, pyruvate dehyd. Interplay: Urea synthesis; gluconeogenesis; fatty acid synthesis, steroid biosynthesis and heme biosynthesis Shuttles: Malate/aspartate, DHAP/glycerol phosphate (electrons) , citrate (acetyl CoA) , carnitine (F.A.s)
1-5 D B C E C - McArdles’ or Cori’s Disease B - Glucose 6-Pi Dehydrogenase Deficiency D - Galactosemia E - Crigler-Najjar Syndrome (bilirubin glucuronyltransferase) A Glucose 6-Pi UDP- Glucouronate Bilirubin Diglucuronide A - Glycogen Storage Disease - Type I
1-5/6 A - Glycogen Storage Disease (Type I): Both GNG and Glycogenolysis HallMarks: Excess Glycogen Accumulation Hypertriglyceridemia Fasting Hypoglycemia Hyperuricemia B - Glucose 6-Pi Dehyd. Def. - Acute Hemolytic Anemia: Stress --> Red Cell [NADPH] H2O2 and Lipid Peroxides MetHb Hemolysis Hematocrit Haptoglobulin Bilrubin/BDG
Glycogen Storage Disease Type I Blood Glucose Feed-Fast Cycles Glucose 6-Phosphate Glycogen PRPP Adipose Lactate DHAP + Acetyl CoA Purines Fatty Acids + Glycerol RBC VLDL
1-6 DHAP Homocysteine A Pyruvate OAA Asp DHAP Cytosolic NADH Utilization: Mitochondrial and Citrate Shuttles Pyruvate --> Lactate Glycerol 3-Pi Synthesis for TAG A.A. Metab.: Ser -->--> 3-Phosphoglycerate A - Homocystinuria, B6 responsive
- B C - + Ketoacidosis (DKA) Acetyl CoA 1-7 Acetyl CoA A - Starvation B - Diabetes (IDDM, Type I) Lipolysis > ketone body utilization C - Carnitine/CAT I Def. Hypoketonemia
C B C - Hyperuricemia - urate underexcretion (renal failure, lactic acidosis, alcoholism) B - Hypoxia or Phosphate Trapping O2 Oxid. Phosp. & ATP ) Pi Oxid. Phosp., Glycolysis, Glycogenolysis PRPP 1-8 A PRPP A - Hyperuricemia/Gout ( HGPRT ) (Lesch-Nyhan Syndrome)
Phosphate Trapping: Fructose Intolerance, Galactosemia, Von Gierke’s Glycogen Storage 1-8 Orotic Aciduria - Poor growth, megaloblastic anemia - unresponsive to folate / B12 PRPP OROTATE A A’ A - Orotate Phosphoribosyl transferase A’ - OMP Decarboxylase Treatment:feed uridine or cytidine UMP Synthase
1-9 Dietary Biotin The Biotin Cycle
1-9 • Only Four Enzymes in Humans Require Biotin: • Pyruvate Carboxylase (mito.) • Propionyl CoA Carboxylase “ • Acetyl CoA Carboxylase (cyto.) • Methyl crotonyl CoA Carboxylase (Leu catabolism) • Many other carboxylases don’t require biotin: • Malic enzyme, PEPCK, Carbamoyl Pi Synthetases, etc. • Biotin/Biotin-like Deficiency: • Biotinidase Deficiency • Holocarboxylase Deficiency • Raw egg ingestion (avidin) Oral biotin supplement often effective
1-10 Multiple Carboxylase Deficiency (Organoaciduria) Methylmalonyl aciduria: enzyme or B12 deficiency , homocysteine may also rise Ketoacidosis - ketoacid of amino acids , anion gap Hypoglycemia and Lactic acidosis - pyruvate carboxylase Dermatitus - fatty acids synthesis - acetyl CoA carboxylase Hyperglycinemia - glycine cleavage enzyme and glycine Hyperammonemia but pH low - N-acetyl Glu synthase , urea cycle
Summary of Multiple Carboxylase Deficiency Mitochondrial propionic acid, branched chain ketoacids and their CoA derivatives Mito. CoA Depletion or inhibition Glycine Cleavage Enzyme Secondary Carnitine deficiency, -oxidation N-acetyl Glu Synthase N-Acetyl Glu Energy Gly NH3 Other Organoacidurias: Maple syrup urine, methylmalonic aciduria and HMGCoA lyase defect act similarly
1-11 Atypical PKU - Biopterin Reductase Deficiency Phenylacetate Phenyllactate Phenylpyruvate B - Albinism Parkinson Disease 5 HO-indole acetic acid Features: Mental retardation despite Phe restriction ; give tetrahydrobiopterin (won’t cross blood brain barrier) - give L-DOPA and 5-HO-Trp Melatonin
Other PKUs: Classical : (> 95 % of PKU’s) Most common inborn error - only degradative pathway involved - restrict Phe to avoid post-natal mental retardation Maternal : Mother is -/+ or -/- for PKU - avoid aspartame (aspartylphenylalanine methyl ester) Serotonin: (CNS) - Neurotransmitter; (Platelet) - vasoconstrictor; (GI) - enterochromaffin cells (tumors) Pheochromocytoma: Cancer of the adrenal medulla. Overproduction of epinephrine - vanillylmandelic high in urine - hypertension
1-12 B Pentoses C B A GLYCOLYSIS 1 * B - GNG Deficiency: Lactic Acidosis / Hypoglycemia A - Heriditary Fructose Intolerance (HFI) C - Galactosemia
1-13 GLYCOLYSIS 2 A A - Hemolytic Anemia - PK deficiency(C.C. 7.8) PKA, ATP
Pyruvate Dehydrogenase 1-14 E3 Deficiency: lactic acid, KG and -ketoacids(bcaa) Five Coenzymes: NAD+, Thiamin, Pantothenic acid, lipoic acid, FAD Five Enzymes: PDH (E1), Lipoyl dehyd. (E2), and transacetylase (E3) kinase and phosphatase
Feasting - Hepatic- High Glucose 1-15 Muscle Uptake, Growth
Fasting - Hepatic -Low Glucose Ala, PKA Ketone Bodies GNG Energy from -oxidation Urea
1-18 Mevinolin, Lipotor - “statins”- lower plasma cholesterol 20-60 % [C30] [C30] A A - Smith-Lemli-Opitz (SLO) syndrome - 7-DHC reductase deficiency - birth defects
METABOLISMINCREASEDECREASE Amino acid degradation Starvation Growth and develop. High Protein Diet Insulin Glucocorticoids Glucagon Glycolysis Insulin Glucagon Gluconeogenesis Glucagon Glucocorticoids Insulin Pentose Pathway Insulin Starvation Carbohydrate Feeding Lipogenesis Low fat diet High Carbohydrate Starvation Lipolysis Glucagon Insulin Cholesterol Biosynthesis Insulin, Glucagon Loss of Bile acids, Starvation, Dietary Cholest. Dietary Cholest. Glycogenesis Insulin Glucagon Glyogenolysis Glucagon Insulin Epinephrine
Metabolic Entry Points of Amino Acids Pyruvate: Trp, Thr, Gly, Ala, Cys, Ser Acetyl CoA/Acetoacetyl CoA (no net carbohyd.synthesis): Ile, Trp, Phe, Leu, Tyr, Lys -Ketoglutarate: Glu, Gln, His, Pro, Arg, Orn Succinyl CoA: Met, Thr, Val, Ile Oxaloacetic Acid: Asp, Asn Fumarate: Asp (urea cycle), Phe, Tyr