Download
1 / 15
320 likes | 819 Views
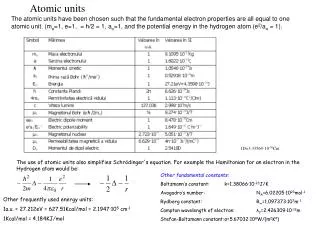
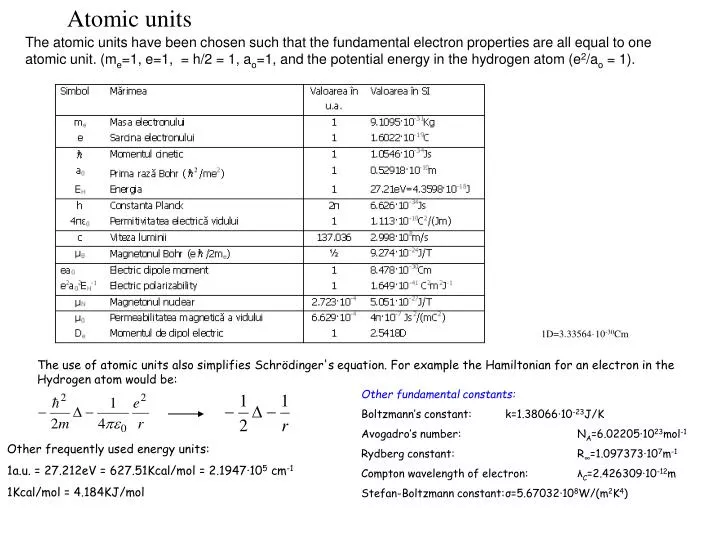
Atomic units. The atomic units have been chosen such that the fundamental electron properties are all equal to one atomic unit. (m e =1, e=1, = h/2 = 1, a o =1, and the potential energy in the hydrogen atom (e 2 /a o = 1). 1D=3.33564 ·10 -30 Cm.

E N D
Atomic units The atomic units have been chosen such that the fundamental electron properties are all equal to one atomic unit. (me=1, e=1, = h/2 = 1, ao=1, and the potential energy in the hydrogen atom (e2/ao = 1). 1D=3.33564·10-30Cm The use of atomic units also simplifies Schrödinger's equation. For example the Hamiltonian for an electron in the Hydrogen atom would be: Other fundamental constants: Boltzmann’s constant: k=1.38066·10-23J/K Avogadro’s number: NA=6.02205·1023mol-1 Rydberg constant: R∞=1.097373·107m-1 Compton wavelength of electron: λC=2.426309·10-12m Stefan-Boltzmann constant: σ=5.67032·108W/(m2K4) Other frequently used energy units: 1a.u. = 27.212eV = 627.51Kcal/mol = 2.1947·105 cm-1 1Kcal/mol = 4.184KJ/mol
Basic concepts, techniques and notations of molecular quantum mechanics • structure of many-electron operators (e.g. Hamiltonian) • form of many-electron wave-functions (Slater determinants, and linear combination of them) • Hartree-Fock (HF) approximation • more sophisticated approaches which use the HF method as a starting point The electronic problem The non-relativistic time-independent Schrödinger equation H|Φ>=E|Φ> H – Hamiltonian operator for a system of nuclei and electrons (1) MA - the ratio of the mass of nucleus A to the mass of an electron ZA – the atomic number of nucleus A Te – the operator for the kinetic energy of the electrons Tn – the operator for the kinetic energy of the nuclei Vee– the operator for the Coulomb attraction between electrons and nuclei Vee – the operator for the repulsion between electrons Vnn – the operator for the repulsion between nuclei A molecular coordinate system (1)– represents the general problem to be separated in two parts: electronic and nuclear problems
Born-Oppenheimer Approximation The nuclei are much heavier than electrons they move much more slowly the nuclei can be considered frozen in a single arrangement (molecular conformation) the electrons can respond almost instantaneously to any change in the nuclear position ► the electrons in a molecule are moving in the field of fixed nuclei ► 2-nd term in (1) can be neglected ► 5-th term in (1) is a constant constant =0 Electronic Hamiltonian describes the motion of N electrons in the field of M point charges (2) Electronic Schrödinger equation: (4) - is the electronic wave-function which describes the motion of the electrons explicitly depends on the electronic coordinatesparametrically depends on the nuclear coordinates parametric dependence the nuclear coordinates do not appear explicitly in Φelec. different wave-function is defined for each nuclear configuration Helec|Φelec>=Eelec|Φelec> (3) Φelec=Φelec({rI};{RA}) (4) Eelec = Eelec({RA}) (5) The total energy: Equations (2) – (6) ≡ electronic problem (6)
If the electronic problem is solved ► we can solve for the motion of the nuclei Since the electrons move much faster than the nuclei ► we can replace the electronic coordinates by their average values (averaged over the electronic wave-function) nuclear Hamiltonian odescribes the motion of the nuclei in the average field of the electrons • nuclear Schrödinger equation Hnucl|Φnnucl> = E|Φnucl> • Φnucl - describes the vibration, rotation and translation of a molecule • E - total energy of the molecule(in the Born-Oppenheimer approximation) • - includes: - electronic energy • - vibrational energy • - rotational energy • - translational energy potential energy surface (PES) Schematic illustration of a potential energy surface The equilibrium conformation of the molecule corresponds to the minimum of the surface
Total wave-function in Born-Oppenheimer approximation: Φ({ri};{RA}) = Φelec({ri};{RA})·Φnucl({RA}) • Born-Oppenheimer approximation • - usually a good approximation • - bad approximation for: • excited states • degenerate or cuasidegenerate states The Antisymmetry or Pauli Exclusion Principle electron spin α(ω) and β(ω) –spin functions (complete and orthonormal) the electron is described by spatial (r) and spin (ω) coordinates: x={r,ω} A many electron wave-function must be antisymmetric with respect to the interchange of the coordinate x (both space and spin) of any two electrons. Φ(x1, x2, ... , xi, ..., xj, ...,xN) = -Φ(x1, x2, ... , xj, ..., xi, ...,xN)
Hartree Approximation (Hartree, 1928) Φi– spin orbitals The form of ΨHPsuggests the independence of Φi Probability density given by ΨHP is equal to the product of monoelectronic probability densities This is true only if each electron is completely independent of the other electrons ΨHP - independent electron model A ♥ A♥ PA=1/13 P♥=1/4 PA♥=1/52=PAP♥ PA is uncorrelated (independent) with P♥. Uncorrelated probabilities Correlated probabilities In a n-electron system of electrons the motions of the electrons is correlated due to the Coulomb repulsion (electron-one will avoid regions of space occupied by electron two). E=εi+εj+…+εn Electronic Hamiltonian can be rewritten: vi is the monoelectronic term of the external potential: Where: In HP, hiwill act only on the wavefunction corresponding to the i-th electron. However, Vee depends on pairs of electrons so that we can not separate the variables in Schrödinger equation. is the monoelectronic operator
Hartree Approximation: the electrons do not interact explicitly with the others, but each electron interacts with the medium potential given by the other electrons Using the variational methods one obtains the energy of the system: where: - core monoelectronic integrals • Coulombian bielectronic integrals • represent the classical repulsion energy between two charge densities described by Φi and Φj Bielectronic potential 1/r12 felt by the electron 1, due to the instantaneous position of electron 2 is replaced by a monoelectronic potential Vi(1) obtained by averaging the interaction between the two electrons over the spatial and spin coordinates of electron 2. Summing over j≠i one obtains the medium potential acting on electron in Φi and which is due to the other N-1 electrons Coulomb operator represents the local medium potential felt by electron 1 and due to the electron described by Φj
Using the Lagrange’s multipliers method Hartree equations: - the energies of molecular orbitals Total electronic energy: In order to find Φi we needΦi SCF procedure SCF procedure in the framework of Hartree approximation
electronic density corresponding to the i-th electron total electronic density Each electron interacts with an electronic density obtained by subtracting its density from the total density Vee potential can be written as: with: gi(r) - interaction energy of the point charge (the considered individual electron) with the other electrons represented as an electronic density Hamiltonian: Hartree equations:
Determinantal wave-functions: Hartree-Fock approximation ΨHP - does not satisfy the Pauli principle - gives a non-zero probability for two electrons to be exactly at the same point in space Fock, Slater, 1930 ΨSD antisimetrized sum of Hartree products with all the possible distributions of the electrons in the molecular orbitals - shorthand notation Using the variational method of Ritz: In Hartree approximation exchange integral
exchange operator: - a non-local operator because its result depends on the value of Φi on entire space and not only on the value of Φi where is located the electron 1 JijKij0 Minimizing the energy by varying the spin orbitals leads to the Hartree-Fock equations: Definig the Fock operator: molecular orbital energies: total electronic energies: total energy: In Hartree approximation
if we use the spatial orbitals: Φ1(x)=φ1(r)α(ω) şi Φ2(x)=φ1(r)β(ω) in the framework of RHF approximation: molecular orbital energies: Hartree-Fock equations = alternative Schrödinger equation in which the exact Hamiltonian has been replaced by an approximate Fock operator - Coulomb operator has been replaced by an operator which describes the interaction of each electron with the average field due to the other electrons
RHF and UHF formalisms Given a set of k orthonormal spatial orbitals (MO) {φi}, i=1,...k 2k spin-orbitals: Φi, i=1,...,2k restricted OM restricted wave-function Restricted wave-function for Li atom But: K1s()2s( )≠0 and K1s()2s()=0 1s() and 1s() electrons will experience different potentials so that it will be more convenient to describe the two kind of electrons by different wave-functions Unrestricted wave-function for Li atom usually, the two sets of spatial orbitals use the same basis set
UHF wave-functions are not eigenfunctions of S2operator !!! spin contamination |2> - exact doublet state |4> - exact quartet state |6> - exact sextet state -approximately a singlet - approximately a doublet For an UHF wave-function, the expectation value of S2 is: where: spin projection procedures (Gaussian)