Download

1 / 47
550 likes | 915 Views
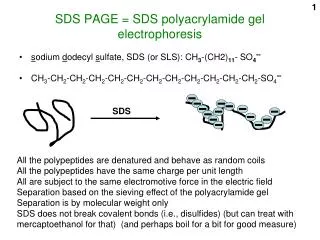
SDS-PAGE and Western Analysis. SDS-PAGE purposes. To separate protein molecules on the basis of molecular weight and To determine the molecular weights of unknowns by comparison to standards. SDS-PAGE preparation of cell extract.

E N D
SDS-PAGE purposes • To separate protein molecules on the basis of molecular weight and • To determine the molecular weights of unknowns by comparison to standards
SDS-PAGE preparation of cell extract • Lyse cells in RIPA buffer containing inhibitors of both proteases and phosphatases. • Centrifuge lysate to remove membranous cellular debris. • Determine protein concentration of the lysate in g/l. • Example: Bradford method - Binding of Coomassie Blue dye by proteins in solution and comparison to standards of known concentration
preparation of cell extract components of the lysis buffer • 50 mM Tris-HCl, pH 8.0 – pH friendly to most proteins • 150 mM NaCl, isotonic saline • Detergents – disrupt lipid bilayers; aid in solubilizing hydrophobic proteins • NP-40 (non-ionic, good solubilization, weakly denaturing) • Deoxycholate (a bile acid, ionic, moderately denaturing) • SDS (synthetic, ionic, excellent solubilization, strongly denaturing)
Components of the lysis buffer (cont’d) • Dithioerythritol – a reducing agent • Prevents inappropriate oxidation of reduced cysteines to disulfide bonds. • Prevents covalent aggregation and precipitation of proteins that are not covalently linked in vivo. • Especially important for native gel electrophoresis. • (See Slide #12. DTT in the lysis buffer is insufficiently concentrated to disrupt the disulfide bonds which form the natural structure of some proteins, but is sufficiently concentrated to prevent inappropriate disulfide bonds from forming.
Components of the lysis buffer (cont’d) • Protease inhibitors • Prevent protein degradation and thereby • allow more accurate determination of molecular weight • Examples • PMSF – inhibits serine proteases • phenylmethanesulfonylfluoride • Leupeptins • tripeptides produced by various species of Actinomycetes • L-leucyl-L-leucyl-Dl-argininal • modified at NH –terminus by acetyl or propionyl • Aprotinin • Found in pancreas and lung, among other tissues • Natural inhibitor of various extra- and intracellular proteases
Components of the lysis buffer (cont’d) • Phosphatase inhibitors • Inhibitors prevent enzymatic removal of phosphates from phosphorylated proteins during extract preparation • Phosphorylated and dephosphorylated proteins migrate differently during SDS-PAGE • Useful information can be gained by knowing whether or not a protein is phosphorylated in vivo in given cells under specific conditions • Examples of general phosphatase inhibitors • NaF • Na3VO4
SDS-PAGE preparation of sample for loading • Major components of the sample loading “buffer” • SDS • DTT • Tracking dye • Glycerol
SDS-PAGEpreparation of sample for loading • Major purposes of boiling in loading buffer are to • denature and coat proteins with SDS • proteins bind SDS with similar ratios of detergent to protein mass • (-) charge on dodecyl sulfate ions ~ = charge/mass ratio for all proteins, so • separation is on the basis of size • reduce disulfide bonds using DTT (or -mercaptoethanol) • Causes disulfide bonded peptides to become independent (see next slide. • Good for determining size of disulfide-bonded subunits
2 2 H H Heat Excess DTT + H
Reduction by monovalent mercaptans -mercaptoethanol Reduction by divalent mercaptans dithioerythritol (reduced) dithioerythritol (oxidized) DTT dithioerythritol
preparation of sample for loading • Dye is included to monitor migration during PAGE • Bromphenol blue • Glycerol is included to make sample denser than running buffer • minimizes diffusion during loading
SDS-PAGE gel system (Note features in red!) • Discontinuous • Two gel layers with different polyacrylamide concentrations • A different buffer for each of the two parts of the gel • Yet a third buffer as the running buffer • Stacking (concentrating) gel • 4% acrylamide (36.5:1, acryl/bis) • 125 mM Tris-H+Cl-, pH 6.8, 0.1% SDS • Resolving (separating) gel • 10% acrylamide (36.5:1, acryl/bis) • 425 mM Tris-H+Cl-, pH 8.8, 0.1% SDS • Running buffer • 25 mM Tris-H+Cl-, pH 8.3, 192 mMglycine, 1% SDS
Why use a discontinuous gel and buffer system for SDS-PAGE? • Purpose of the stacking gel: to concentrate all the proteins in the sample into a thin band at the top of the resolving gel • Makes it possible to use a dilute sample • Purpose of the resolving gel: to separate the proteins on the basis of size. • The next set of slides will address how the stack works. • Following that will be a set of slides on the resolving system.
stacking gel • Concentrates proteins because it • Has large pores (4%), so proteins of all sizes • move easily through the pores of the stacking gel until they meet a frictional barrier at the top of the resolving gel, with its smaller pores (10%). But that’s not the only way the proteins are concentrated! Clever design of a discontinuous buffer system increases the concentrating effect of the stacking gel on the proteins in the sample!
stacking gel • Concentrates proteins also because it • uses the stacking and running buffers to form a voltage gradient protein mobility • Stacking gel buffer is of • Low salt concentration • Cl- = leading ion because it is small, negatively charged, and moves quickly through gel • Running buffer ion is primarily • glycine = trailing “ion”, which at pH 6.8 is nearly neutral • A region of low ionic strength quickly develops between Cl- and glycine, generating a voltage gradient. • Large, negatively charged proteins are left to constitute most of the molecular current, and move quickly to the bottom of the stacking gel.
pH 6.8, neutral Notice the progression of Cl-, negatively charged proteins of different sizes (P-), and mostly neutral glycine (G) in the next three slides.
G G G G G P-- P- P- P- Cl- P- Cl- P-- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl-
G G G G G P-- P- P- P- P- P-- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl-
G G G G G P-- P- P-- P- P- P- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl-
G G G G G G G P-- P- P-- P- P- P- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl-
On to the resolving gel . . . • The resolving gel separates proteins as a function of • percentage acrylamide • ratio of acrylamide to bis • extent of difference in size between the proteins being resolved
The resolving gel • Has a higher [ion] (0.425 M Tris-H+Cl-) than the stacking gel (0.125 M Tris-H+Cl-) so • Proteins contribute less to the total ionic current than they did in the stacking gel, and as a result the • mobilities of proteins and differences in mobilities among proteins of different sizes become more apparent.
Resolving gel • In addition, the resolving gel has a higher pH (8.8) than the stacking gel (6.8) so glycine • takes on a more negative charge, thereby the increasing the total ion concentration and • because it is small, moves ahead of the proteins. • So proteins move slowly through the gel and are resolved by friction on the basis of size.
H2 pH 6.8, neutral pH 8.8, negatively charged
G G G G G G G P-- P-- Cl- Cl- Cl- Cl- Cl- P- P- P- P- Cl- Cl- Cl- Cl- Cl- Cl- G- Cl- Cl- G- Cl- Cl- Cl- Cl- Cl- G- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- G- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl-
G G G G G G G G G G Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- G- Cl- Cl- G- P-- G- Cl- Cl- Cl- Cl- P-- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- P- P- Cl- Cl- P- P- Cl- Cl- Cl- Cl- Cl- Cl- Cl- G- Cl- Cl- Cl- Cl- G- Cl- Cl- Cl- Cl- Cl- G- Cl- Cl- Cl- Cl- Cl-
Immunoblotting • Electrophoretic transfer is used to move proteins out of gel onto membrane in equivalent relative positions for subsequent antibody probing • Nitrocellulose membrane is most commonly used • Nature of bonding between proteins and nitrocellulose is not known. • Other membrane types can be used • PVDF, nylon, charged nylon • Each has advantages and disadvantages • Electrophoretic transfer • Fast • Usually complete
Immunoblotting • Equilibration of gel in transfer buffer (25 mM Tris, 192 mM glycine, 20% v/v methanol) • Removes excess electrophoresis buffer salts from gel. • The higher the ionic strength during electrophoretic transfer, the more heat is generated. • Excess heat is a safety hazard.
Immunoblotting • Equilibration of gel in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) • Removes detergents from gel. • Detergents interfere with binding of protein to membrane. • 0.01 - 0.02% SDS in transfer buffer can improve transfer out of gel. • Equilibrates the gel with methanol. • Methanol is required for binding of proteins to nitrocellulose. • Methanol shrinks the gel. • Gel must be allowed to complete shrinking before blot sandwich is assembled. • Shrinkage may interfere with transfer of larger proteins.
After transfer • Check for success of transfer • See if the colored protein standards are on the membrane, and/or • Stain the proteins on the blot with stain that does not interfere with immunological probing. • Example: Ponceau S • Can also be done after immunoprobing
Immunoprobing • Choice of blocking agents • Choice of 1o Antibody • Choice of 2o Antibody • Choice of antibody concentrations • Visualization system
Factors to Consider when Choosing the Type of 1o Antibody Preparation to Use in a Western
Immunoprobing • Choice of 2o Antibody • 2o Ab must recognize the common portion of the 1o Ab. • 1o Ab is raised in a given animal • e.g., mouse • i.e., mouse was immunized by injection with the antigen • e.g., denatured p53 • 2o Ab must be raised in a different animal • e.g., goat or rabbit • i.e., animal immunized by injection with Ig that is of the • same class (IgG) as 1o Ab , but of different irrelevant specificities
Immunoprobing • Choice of antibody concentrations • Use as little 1o and 2o Ab as possible to generate detectable signal. Minimization of [Ab] • cleaner results • lower cost • Too much of either 1o or 2o Ab • background • non-specific binding • Various 1o and 2o Ab concentrations must be tried to • optimize signal • minimize noise
Western Immunoprobing • Enzymatic detection systems • Alkaline phosphatase • Horseradish peroxidase • Direct vs. indirect detection • Note, the meanings of direct vs. indirect in immunoprobing on a blot is NOT the same as for an ELISA.
Immunoprobing • Enzymatic detection systems • Horseradish peroxidase catalyzes the rx . . • Luminol + H2O2 light + 3-aminophthalate + N2 + H2O
Western Immunoprobing • Direct detection - 1o Ab is enzyme-conjugated • Fewer background bands and background than indirect detection, but • Less sensitive and • Requires that 1o Ab of every different specificity be purified and conjugated to enzyme • Indirect detection - 2o Ab is enzyme-conjugated • More sensitive than direct • Less expensive than direct - one preparation of labeled 2o Ab can recognize all 1o Abs from the same species. • HRP conjugated rabbit anti-mouse IgG can be used in a Western for any protein recognized by a 1o Ab raised in mouse.
Comparing your Western positive band to size standards • Make a standard curve on semi-log paper,plotting the distance traveled against the known sizes of the standards. • Measure the distance traveled by the protein giving you a positive band on your Western. • Determine the size of your protein from the standard plot.
Myosin, 202 kd MWs adjusted to account for masses of covalently bound dyes -galactosidase, 133 kd (red) BSA, 71 kd Carbonic anhydrase, 41.8 kd Soybean trypsin inhibitor, 30.6 kd Lysozyme, 17.8 kd Aprotinin, 6.9 kd
Predict the distance traveled by p53 in each of the two gels for which the standards are plotted at right. 7% resolving gel 10% resolving gel = distance traveled