Download

1 / 24
240 likes | 327 Views
Team WALA – Weeks 4 & 5. Team: Will Darby, Alfredo Lopez, Len Russo, Alan Levin (Team Leader) Application: Gene Expression in Human Breast Cancer Data Investigations: Detailed investigations into k-means clustering with feature selected datasets

E N D
Team WALA – Weeks 4 & 5 • Team: • Will Darby, Alfredo Lopez, Len Russo, Alan Levin (Team Leader) • Application: • Gene Expression in Human Breast Cancer Data • Investigations: • Detailed investigations into k-means clustering with feature selected datasets • Working with the clinical augmentation to the breast cancer dataset
Overview • Our team worked with the Human Breast Cancer dataset for the entire duration of our project. We originally started out learning how to use the tools available in Weka and trying to understand which tool provided the best classification accuracy and under what conditions and variables. We investigated boosting, bagging, support vector machines and random forests. We initially used five-fold validation to test the various tools. We spent the most time working with random forests and support vector machines. Next we moved on to understanding the CFS/Best First Search and Infogain/Ranker feature selection methodologies of Weka. The team also looked into ROC curves. We then worked with 2-way hierarchical clustering and SAM analysis in TMeV. The team also spent some time looking into a genetic algorithm approach to classifying the data. The team spent the most time working with SimpleKMeans clustering in Weka and k-means clustering in TMeV. Finally the team spent some time looking into the Clinical data enhancement to the Human Breast Cancer dataset. • The most interesting thing that the team discovered was that SimpleKMeans and k-means could produce accuracies comparable to those of the most complex classification and clustering methods. This discovery suggested to us that we investigate these clustering methods in more detail.
General Results • We determined that the best accuracies from random forests were gained when we used hundreds of trees and dozens of randomly selected features. • We also realized that the classification problem could be described as a matrix multiplication problem This lead to one of two methods we came up with on how to use genetic algorithms to solve the classification problem. We decided not to implement a genetic algorithm – but the descriptions of these algorithms are available on the team’s Wiki page under Week 3 results. • We also carried out some additional analysis into Random Forest accuracies. The best we found was 87.9%. • Once we started using feature selection methodologies we realized that many fewer than the 1554 genes in the dataset were sufficient to obtain excellent accuracies.
SimpleKMeans becomes Interesting • We saw that with k=8 that one of the “extra” classes was actually almost 100% Basal-like and that a large number of misclassifications were Luminal-A classified as Luminal-B and vice versa. • We next proceeded to see how the various feature selection methods – CFS/BFS, Infogain/Ranker and SAM – would impact clustering accuracies.
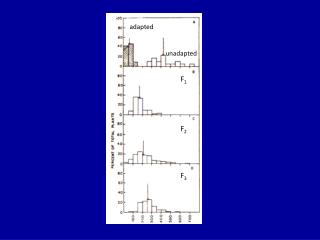
Detailed Results of k-means Investigations • Our approach was to create the following datasets: • CFS/BFS: 138 genes • SAM: 236 genes (238 minus 2 duplicates) • Infogain/Ranker/250: 250 genes • Infogain/Ranker/300: 298 genes (300 minus 2 duplicates) • Overlapped dataset: 233 genes • Any gene that was present in at least 2 of the other datasets • We found that the datasets overlapped as follows: • 84% of the SAM genes are matched by Infogain300 genes • 62% of the CFS genes are matched by Infogain300 genes • 49% of the CFS genes are matched by SAM genes
Detailed Results of k-means Investigations • We created a number of tables showing how different datasets were clustered using TMeV’s k-means clustering while varying clusters from k=6 and k=10. • [Please see the datasets posted on the team’s Wiki page under Week 4 results.] • We discovered the following percentage errors when k=6: • CFS/BFS: 11.2% SAM: 25.9% • Infogain/Ranker/250: 20.9% Infogain/Ranker/350: 22.8% • Overlapped dataset: 15.9% • These results were obtained using Weka’s SimpleKMeans: • CFS/BFS with k=6 and seed =2: 11.6% • [The difference between k-means and SimpleKMeans was that k-means didn’t find a cluster to assign Luminal B to – it grouped it with Luminal A.]
Detailed Results of k-means Investigations • While the CFS/BFS dataset had provided us with an accuracy of 88.8% - better than most other methodologies – our expectations were that 138 genes weren’t enough to get the best results and therefore some “augmented” dataset should be more accurate. • We weren’t able to create that dataset with the overlap dataset – so we took another approach. • We split the original dataset in half: • Normal breast-like & Claudin-low & Basal-like • Luminal-A & Luminal-B & HER2+/ER-
Detailed Results of k-means Investigations • We ran both datasets through CFS/BFS feature selection and then clustered those datasets using SimpleKMeans with k=3 • We obtained the following results: • Split dataset #1 with CFS and SimpleKMeans k=3 and seeds=8/9 97.9% • Split dataset #12 with CFS and SimpleKMeans k=3 and seed=9 91.2% • We then tried merging the CFS/BFS datasets from the 2 halves and that dataset with the original CFS/BFS dataset and obtained the following results: • 3x CFS individual sets SimpleKMeans k=6 and seed=2 88.8% • 2x CFS individual sets SimpleKMeans k=6 and seed=2 87.5% • While we did obtain the same 88.8% result, it was not nearly as good as we would have expected.
Biology gets in the Way! • Our team also read papers on gene modules and tried to use the GRAM algorithm - http://psrg.lcs.mit.edu/GRAM/Index.html • Unfortunately after expending a lot of effort we weren’t able to figure out how to access transcription site or gene module data for our dataset(s) and could not proceed. • Some of our lessons learned from this activity: • Advanced Bioinformatics relies on thorough subject matter knowledge. • e.g. Georg Gerber first received an M.P.H. in Infectious Diseases from Berkeley. • No longer about processing ‘data’ – instead uses ‘expert’ information to aid algorithms. • Finding appropriate data sources is difficult • Expression data referred to ‘well-known’ datasets • Public regulator data is highly specialize, e.g Saccharomyces cerevisiae • Another problem that our lack of genetics background caused was our inability to understand why the various small datasets that we created with CFS/BFS were so good at focusing k-means clustering.
Clustering and augmenting the original dataset with clinical details • The team did spend some time trying to determine if there were any correlations between the clinical details and clusters that were created. • We investigated good (>5 years) & bad prognosis (<5 years) • We investigated Alive versus Dead
K-Means/K-Medians Sample Selection Distance Metric Selection Parameters Hierarchical Clustering Tree Selection Distance Metric Selection Linkage Method Selection Cluster Samples Euclidean Distance Calculate K-Means Number of Clusters: 6-12 Maximun Iterations: 50 Construct Hierarchical Tree Gene Tree, Sample Tree Euclidean Distance Average linkage clustering How to TMeV Clustering Results
How to TMeV Clustering Results • Analysis Results • KMC – samples (x) • Tables views • For each Cluster, Count Sample Name, and its frequency • From any Cluster result, calculate Classes frequency • Choose most accurate n-Clusters Result • Per each Class, and from most accurate n-Cluster Result, calculate Accuracy per Cluster/per Sample • Change Display/Sample Column Label when necessary to figure out different variables
Kaplan-Meier Survival Analysis • Merged Original Gene Expression Data with Clinical Data • Summary statistics: • Total observed: 160 • Total failed: 43 • Total censored: 117 • Mean survival time: • Mean: 64.999 • Standard Dev: 4.073 • Lower bound (95%): 57.017 • Upper bound (95%): 72.981
Kaplan-Meier Recurrence Analysis • Summary statistics: • Total observed: 161 • Total failed: 57 • Total censored: 104 • Mean relapse time: • Mean: 59.453 • Standard Dev: 4.771 • Lower bound (95%): 50.103 • Upper bound (95%): 68.803
Possible Next Steps and Conclusions • It could be possible to work with the clusters of genes created using TMeV’s 2-way hierarchical clustering. • Eliminating a cluster at a time and seeing the effects on k-means clustering could show how each cluster impacts each of the 6 classes. • We were surprised that Weka’s SimpleKMeans and TMeV’s k-means did so well in clustering the dataset – particularly when using CFS/BFS feature selection. • We were disappointed that our lack of detailed knowledge about genetics kept us from understanding why our best feature-selected datasets performed the way they did.
Feature Selection • Team WALA has used three primary methods of feature selection: CFS/BFS, InfoGain/Ranker and SAM. All are heuristic approaches for selecting features sometimes containing “fudge factors”. • We would like to rely on heuristic “filters” as doing an exhaustive wrapper search is very time consuming. • CFS/BFS is a correlation based heuristic. • CFS/BFS finds those features best correlated to the class and least correlated to each other. • InfoGain/Ranker is based on entropy. • This is similar to using information gain in tree classifiers on individual attributes. • For any subset, we may add the attribute yielding the highest information gain. • Once attributes have been ranked, we may set a threshold picking a subset with highest information gain. • SAM (Significance Analysis of Micro-array) is a heuristic feature selector primarily used for micro-array data. • Pairs of genes are differenced and compared to their joint standard deviation plus a fudge factor. • Differences above a threshold are deemed significant resulting in adding an attribute. This is thus a T-test type heuristic.
Mathematical Description of the Human Breast Cancer Classification Problem • We are trying to solve: A*b = c where A is 232x1544, b is 1544x1 and c is 232x1. • We know A (the data matrix) and c (the class assignment) = [0 to 5] for each exemplar. • If we knew b - the weight matrix - we would be done. But the problem is very ill-conditioned. • We can formulate a least squares problem: A’*A*b = A’*c b = (A’*A)-1A’*c • A’*A determines the difficulty of the problem and is 232x232 • It seems reasonable that less than 250 features should be required for a classifier.
Variation with Seeds of Cross-Validation Scores for Random Forests on Breast Cancer Data • Results for 5-fold cross-validation shown right. Seeds were varied from 1, 100, 1000. • We see the deviations overlap; hence, there is a high confidence these results are statistically the same. • However, mean scores vary by several per cent. • Best score was 87.9%
Variation of Scores with Attributes and Number of Trees • These images are the result of 100 runs each with different seed for each image. Each image shows variation with number of attributes and number of trees. Trees vary from 70-290, attributes from 35-125. • Scores in two of three (Seed = 1, 1000) seem to be high for a specific number of attributes. This number is ~2x or ~3x sqrt(Nattributes). For Seed=100, no trend is apparent. The maximum score is the same in two of three runs. • It is clear that maximum score should be taken with a grain of salt. Result can vary with seed. See summary preceding summary.