Download
1 / 78
790 likes | 1.39k Views
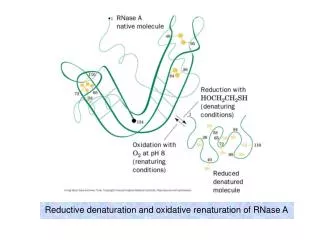
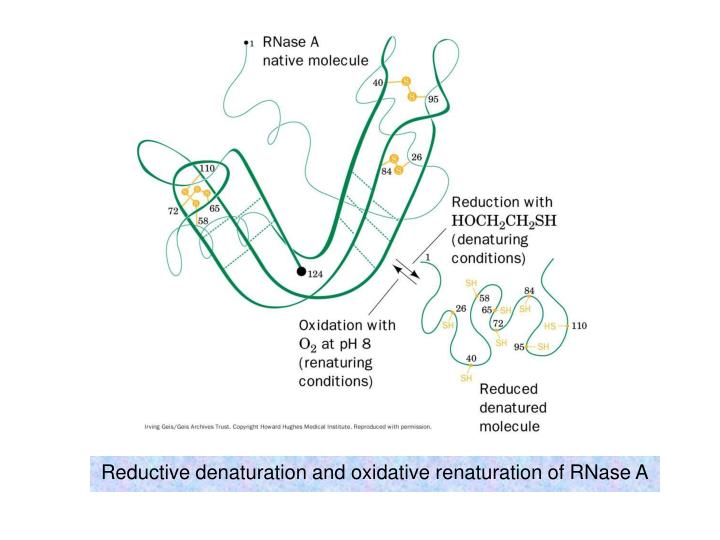
Reductive denaturation and oxidative renaturation of RNase A. Plausible mechanism for the thiol- or enzyme-catalyzed disulfide interchange reaction in a protein. protein disulfide isomerase. C-chain needed to direct proper disulfide bond formation. Primary structure of porcine proinsulin.

E N D
Reductive denaturation and oxidative renaturation of RNase A
Plausible mechanism for the thiol- or enzyme-catalyzed disulfide interchange reaction in a protein protein disulfide isomerase
C-chain needed to direct proper disulfide bond formation Primary structure of porcine proinsulin
Determinants of Protein Folding A. helices/sheets predominate in proteins because they fill space efficiently B. protein folding is directed mainly by internal residues (protein folding is driven by hydrophobic forces) C. protein structures are organized hierarchically
Determinants of Protein Folding (cont.) D. protein structures are highly adaptable E. secondary structure can be context-dependent
“chameleon” sequence: AWTVEKAFKTF (unfolded free in solution) NMR structure of protein GB1
Determinants of Protein Folding (cont.) F. dependence of protein fold on primary sequence
created by changing 50% of the 56 residues in GB1 not all residues have equally important roles in specifying a specific fold X-ray structure of Rop protein, a homodimer of aa motifs that associate to form a 4-helix bundle
The Levinthal Paradox 2n backbone torsions, n-residue protein: ~10n structures time to explore all structures: t = 10n/1013 s-1 for a 100-residue protein: t = 1087 s Conclusion: proteins fold via an ordered pathway or set of pathways
Experimental Methods to Monitor Protein Folding
(UV/ VIS/ fluorescence /CD) cold denatured proteins / T-jump A stopped-flow device: 40 ms dead-times
molar extinction coefficient UV absorbance spectra of the three aromatic amino acids, phenylalanine, tryptophan, and tyrosine
eL and eR differ (circularly polarized light); measure ∆e Circular dichroism (CD) spectra of polypeptides
Pulsed H/D Exchange X-H + D2OX-D + HOD Used to follow the time course of protein folding by 2D NMR a. Denatured protein in D2O b. Dilute with H2O and allow to fold for time tf c. Increase pH to initiate D-H exchange (10-40 ms) d. Lower pH; allow to completely fold e. Determine which amide protons are protonated and deuterated
Landscape Theory of Protein Folding Polypeptides fold via a series of conformational adjustments that reduce their free energy and entropy until the native state is reached. There is no single pathway or closely related set of pathways that a polypeptide must follow in folding to its native state. The sequence information specifying a particular fold is both distributed throughout the polypeptide chain and highly overdetermined.
Closer mimic of an actual folding pathway Folding funnels: Rugged energy surface
Folds via an ordered pathway: involves well defined intermediates Polypeptide backbone and disulfide bonds of native BPTI (58 residues, three disulfide bonds)
Renaturation of BPTI: protein primary structures evolved to specify efficient folding pathways as well as stable native conformations
Folding accessory proteins A. Protein disulfide isomerases (PDI) B. Peptidyl prolyl cis-trans isomerases C. Molecular chaperones
A folding accessory protein Reactions catalyzed by protein disulfide isomerase (PDI). (a) Reduced PDI catalyzes the rearrangement of the non-native disulfide bonds.
Reactions catalyzed by protein disulfide isomerase (PDI). (b) The oxidized PDI-dependent synthesis of disulfide bonds in proteins.
homodimer; eukaryotic; each subunit consists of four domains Cys 36 and 39 exposed NMR structure of the a domain of human protein disulfide isomerase (PDI-a) in its oxidized form. (a) The polypeptide backbone is shown in ribbon form.
Cys 36 is located in a hydrophobic patch Oxidized PDI-a is less stable than reduced PDI-a NMR structure of the a domain of human protein disulfide isomerase (PDI-a) in its oxidized form. (b) The molecular structure as viewed from the bottom.
Peptidyl Prolyl Cis-Trans Isomerases (PPIs) Xaa-Pro peptide bonds: ~10% cis PPIs catalyze the otherwise slow interconversion of Xaa-Pro peptide bonds between their cis and trans conformations; accelerate the folding of Pro-containing polypeptides. Two families: cyclophilins and FKBP12 (based on known inhibitors)
Unfolded proteins in vivo have a great tendency to form intramolecular and intermolecular aggregates. Molecular chaperones prevent/reverse improper associations, especially in multidomain and multisubunit proteins. Function by binding solvent-exposed hydrophobic surfaces reversibly to promote proper folding Many chaperones are ATPases.
Classes of Chaperones A. Heat shock proteins 70: 70 kD monomeric proteins B. Chaperonins: form large multisubunit cage-like assemblies C. Hsp90: involved in signal transduction; very abundant in eukaryotes D. Nucleoplasmins: acidic nuclear proteins involved in nucleosome assembly
Chaperonins GroEL/ES system 14 identical ~60 kD subunits in two rings; creates a central cavity Electron micrograph-derived 3D image of the Hsp60 (GroEL) chaperonin from the photosynthetic bacterium Rhodobacter sphaeroides.
X-ray structure of GroEL. (a) Side view perpendicular to the 7-fold axis.
X-ray structure of GroEL. (b) Top view along the 7-fold axis.
cis ring trans ring X-ray structure of the GroEL-GroES-(ADP)7 complex.
bound ADP shown in cis ring X-ray structure of the GroEL-GroES-(ADP)7 complex.
apical intermediate equatorial Domain movements in GroEL. (a) Ribbon diagram of a single subunit of GroEL in the X-ray structure of GroEL alone.
Domain movements in GroEL. (b) A GroEL subunit in the X-ray structure of GroEL-GroES-(ADP)7.
Domain movements in GroEL. (c) Schematic diagram indicating the conformational changes in GroEL when it binds GroES.
Apical domain of GroEL in complex with a tight-binding 12-residue polypeptide (SWMTTPWGFLHP).
(a) (b) Movements of the polypeptide-binding helices of GroEL.
Reaction cycle of the GroEL/ES chaperonin system in protein folding.
Models for GroEL/ES Action A. Anfinsen cage model: folding within complex B. Interative annealing: reversible release of partially folded intermediates
Refolding of RiBisCO; requires assistance to reach native state black: no components red: released only after native state is reached green: released after one turnover Rate of hydrogen-tritium exchange of tritiated RuBisCO.
exchange with solvent Schematic diagram of the mechanism of stretch-induced hydrogen exchange by the GroEL/ES system.
Protein Structure Prediction Secondary structure a) Chou-Fasman method Frequency at which a given aa occurs in an a helix in a set of protein structures = fa = na/n, where na = number of amino acid residues of the given type that occur in a helices, and n = total number of residues of this type in the protein set Propensity of a particular aa residue to occur in an a helix = Pa = fa/<fa>, where <fa> is the average value of fa for all 20 residues Pa > 1: residue occurs with greater than average frequency in an a helix Also applies to b-structure
H = strong former h = former I = weak former i = indifferent b = breaker B = strong breaker Propensities and classifications of amino acid residues for a helical and b sheet conformations.
B. Reverse turns: Rose method Occur on the surface of a protein; locations of minimal hydropathy (exclude helical regions)
Rationale for Observed Propensities For a-helix: appears related to the amount of side-chain hydrophobic surface buried in the protein For Pro: low a propensity caused by strain For Gly: low a propensity caused by reduced entropy and lack of hydrophobic stabilization For Ala: high a propensity caused by lack of a g substituent; reduced entropic cost; minimal hydrophobic stabilization