Download

1 / 19
190 likes | 332 Views
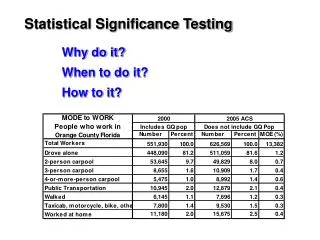
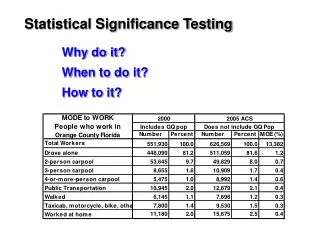
Statistics Applied to Bioinformatics. Significance testing. Compare target score with rest of scores. Score 330 lies here, way outside the heap of random scores. freq. The scores of unrelated (random) database hits. score. Example: scanning a database with a sequence

E N D
Statistics Applied to Bioinformatics Significance testing Jacques van HeldenJacques.van.Helden@ulb.ac.be
Compare target score with rest of scores Score 330 lies here,way outside the heap ofrandom scores freq The scores of unrelated (random) database hits score • Example: scanning a database with a sequence • The query sequence is successively compared with each database entry, and a score is assigned for each comparison • The best match returns a score of 330 • The score distribution for all the database entries is provided • How significant is this match ? slide from Lorenz Wernisch
Approach • We will first fit a normal distribution over the data • Which parameters do we need to fit a normal distribution over a data set ? • This fitted curve will be used to estimate the significance of this score • How do we estimate the significance of the score ?
Fit a Normal (Gaussian) distribution score s = 330 m= -47.1 s = 20.8 slide from Lorenz Wernisch
p-value for Normal distribution 0.0117 • The red area is theprobability for a random normal distribution N(-47.1,20.8) to give a score > 0 • Pr[s > 0] = 0.0117 > pnorm(330,-47.1,20.8, lower.tail=F) 9.27032e-74 adapted from Lorenz Wernisch
P-value and expected matches • In the previous slide, we saw that P(X > 0) = 0.0117 • Let us assume that the database contains 200,000 sequences • If we set the threshold to 0, how many matches would be expect at random ?
From P-value to E-value • If p=P(X >0)=0.0117 and the database contains N=200,000 entries, we expect to obtain N*p = 2340 false positives ! • We are in a situation of multi-testing : each analysis amounts to test N hypotheses. • The E-value (expected value) allows to take this effect into account : • E-value = P-value * N • Instead of setting a threshold on the P-value, we should set a threshold on the E-value. • If we want to avoid false positive, this threshold should always be negative. • Threshold(E) 1 • This is equivalent to Bonferoni's rule • In case of multi-testing, the threshold on P-value should be adapted to the number of tests • Threshold(P) 1/N
Significance testing • We can evaluate the significance of each observation, by calculating its P-value. • Under the assumption of normality, the P-value can be obtained from z-scores. Z-scores represent the number of standard deviations from the mean. P-value x
Statistics Applied to Bioinformatics Multi-testing corrections Jacques van HeldenJacques.van.Helden@ulb.ac.be
Bonferoni rule • Multi-testing • Assessing the significance of each gene on a chip represents thousands of simultaneous tests. Let N be the number of genes. • The risk of error (P-value) associated to each gene will thus be challenged N times. • The significance thresholds used for single testing (0.01, 0.001) are thus likely to return many false positive. • Bonferoni rule • Adapt the threshold to the number of simultaneous tests.
E-value • An alternative but equivalent way to treat the problem of multi-testing is to calculate the expected value for each observation. • One can then choose the E-value according to the number of false positive considered as acceptable.
Family-wise Error Rate (FWER) • Another correction for multiple testing consists in estimating the probability to observe at least one false positive in the whole set of tests. This probability can be calculated quite easily from the P-value (Pval).
False Discovery Rate (FDR) • Yet another approach is to consider, for a given threshold on P-value, the False Discovery Rate, i.e. the proportion of false predictions within a set of predictions.
Summary - Multi-testing corrections • Bonferoni rule adapt significance threshold • E-value expected number of false positives • FWER Family-wise error rate: probability to observe at least one false positive • FDR False discovery rate: estimated rate of false positives among the predictions
Statistics Applied to Bioinformatics Exercises - Significance testing Jacques van HeldenJacques.van.Helden@ulb.ac.be
Exercise - GGCGCC in the genome of E.coli • The genome of Escherichia coli (4,639,221 base pairs) contains 94 occurrences of the hexanucleotide GGCGCC. • Knowing that this genome contains 50.78% of G/C • what would be the probability to find a match at any position (with a Bernouilli model) • how many occurrences would be expected at random ? • assess the significance of the observed number of occurrences of GGCGCC ?
Exercise - motif in upstream sequences • Hexanucleotide occurrences were counted on both strands, in 800bp upstream sequences of • A set of 6 nitrogen-regulated genes • The complete set of 6,448 genes of the yeast genome • The motif GATAAG has the following occurrences • 24 occurrences for the 6 nitrogen regulated genes • 2,763 occurrences in the complete set of upstream sequences • Questions • How many occurrences would be expected at random ? • What is the significance of the observed number of occurrences ?
Statistics Applied to Bioinformatics Additional material Jacques van HeldenJacques.van.Helden@ulb.ac.be
Filtering genes on the basis of their log-ratio in microarray data • In the first publications on microarray analysis, genes were filtered on the basis of a threshold on the log-ratio. Typically, papers from Stanford were considering as significantly regulated all genes with • R/G log2(R/G) regulation • 2 1 up-regulated • 1/2 -1 down-regulated • These thresholds were based on an empirical observation (a control chip). They however suffer from several drawbacks • They do not rely on any statistical or probabilistic criterion. • They do not take into account the bias in centring. This can be circumvented by first centring each chip independently. • They do not take into account the chip-specific dispersion. Among a series, some chips may have a wider dispersion than others, due to experimental bias (scanner setting, problems with dye, ...). • A scaling is thus required, but after scaling, the values do not directly represent expression ratios anymore.