Download
1 / 62
630 likes | 882 Views
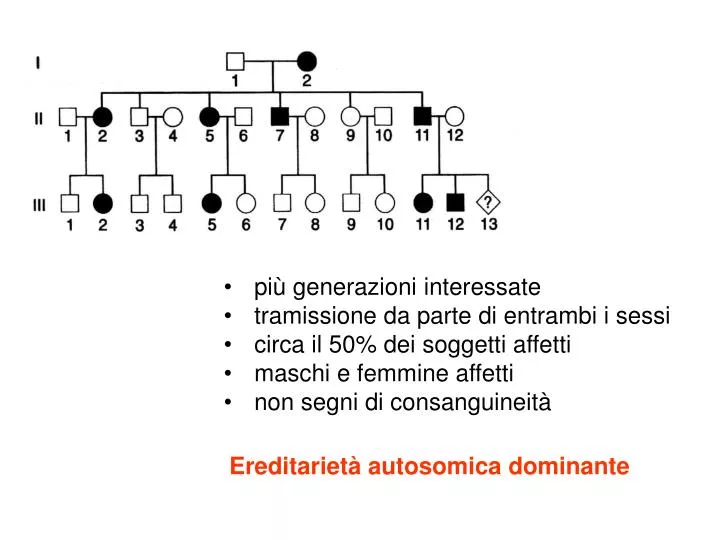
più generazioni interessate tramissione da parte di entrambi i sessi circa il 50% dei soggetti affetti maschi e femmine affetti non segni di consanguineità. Ereditarietà autosomica dominante. Trasmissione ereditaria dei caratteri mendeliani. Dominante.

E N D
più generazioni interessate • tramissione da parte di entrambi i sessi • circa il 50% dei soggetti affetti • maschi e femmine affetti • non segni di consanguineità Ereditarietà autosomica dominante
Trasmissione ereditaria dei caratteri mendeliani Dominante - ogni tratto o carattere che si esprima nell’ eterozigote - nel caso di malattia: una sola copia del gene mutato è sufficiente per esprimere il fenotipo affetto Genotipo : eterozigote Fenotipo : affetto Codominante: l’eterozigote esprime un fenotipo distinto da quello dei due stati omozigoti per es. gruppi sanguigni, enzimi eritrocitari, etc.
Eredità autosomica dominante Se un genitore è normale (bb) e l’altro è affetto (Bb) da una malattia autosomica dominante, il 50% dei figli sarà eterozigote affetto (Bb), ed il 50% sarà omozigote normale (bb). Nelle malattie autosomiche dominanti, la lettera maiuscola indica il gene che dà la malattia: un solo allele mutato dà origine al fenotipo affetto Rischio di ricorrenza = 50% (0.5)
Eredità autosomica dominante Se entrambi i genitori sono affetti (Bb) da una malattia autosomica dominante, allora il 75% dei figli sarà affetto (BB o Bb), e il 25% sarà omozigote normale (bb). Nota:è raro che un individuo affetto da una malattia autosomica dominante sia omozigote per il gene che dà la malattia.
Eredità autosomica dominante • si manifesta in uguale misura nei due sessi • la trasmissione non dipende dal sesso • il figlio di un genitore affetto e di uno sano ha il 50% di probabilità di essere affetto • un individuo affetto ha comunemente un genitore affetto • …………….mutazioni de novo • individui non affetti non trasmettono la malattia • ….……penetranza incompleta
III-1 III-2 Carattere AD a penetranza completa tasso m di “ nuove mutazioni ” R = 0.5 (50%) Carattere AD a penetranza incompleta (penetranza del 70%) R (III-1) = 0.5 ??? R (IV-1) = 0.25 ???
III-1 III-2 Ipotesi 1Ipotesi 2 PortatoreNon Portatore 1 / 2 1 / 2 prior p. 3 / 10 1 conditional p. ½ x 3/10= 3/20 ½ x 1=1/2joint p. 3/201/2 3/20 + 1/21/2 + 3/20posterior p. = 3/13 (23%) = 10/13 (77%) Teorema di Bayes: Rischio di ricorrenza III-1 R (III-1) di trasmettere = ½ x 3/13 = 3/26(11.5%) R (III-1) di avere un figlio affetto = 3/26 x 7/10 = 8%
spermatogenesi oogenesi Nuova mutazione mosaicismo Mutazioni nuove o “de novo” identificate nelle malattie dominanti o recessive X-linked, molto raramente producono malattie autosomiche recessive (entrambe le copie del gene devono essere mutate) il rischio di ricorrenza è basso ma leggermente superiore all’incidenza della malattia nella popolazione a causa del mosaicismo della linea germinale
Aspetti da considerare nelle malattie AD • Penetranza - completa – incompleta - diversa per fasce di età • Espressività - presenza – assenza - espressività variabile • Tasso di nuove mutazioni casi sporadici – famigliari • Percentuale di mosaicismi germinali
penetranza >90% Ipotesi: mosaicismo germinale o somatico espressività variabile Retinoblastoma (1:15.000-20.000) 60% dei casi 40% dei casi Rb Rb Rb Rb bilaterale Rb multifocale monolaterale Retinoblastoma sporadico monolaterale unico
Retinoblastoma • f 1 : 15.000-20.000 nati • 60% dei casi: tumore monolaterale unico • inattivazione somatica di Rb1 (-/-) • 40% dei casi: tumore multifocale e/o bilaterale • con storia famigliare POSITIVA • trasmissione AD di mutazione Rb1 (-/+) : • inattivazione somatica del secondo allele (-/-) • con storia famigliare NEGATIVA • mutazione de novo in un gamete (maschile) • mosaicismo germinale in un genitore (6-10%) • mosaicismo somatico nell’affetto (10%) RR=0.5, RT=0.5 RR=??, RT=??
Gene Rb1 • 13q14: 27 esoni, mRNA 4,840 , proteina 928 aa • più di 400 mutazioni note • 85-90% mutazioni troncanti (NS, FS, SS): esoni 2-25
SCA : • che tipo di ereditarietà • qual’è la probabilità di essere • portatore per il padre e per la • probanda • è possibile eseguire un test • genetico ? A che scopo ? • a chi offrirlo
Ipotesi 1Ipotesi 2 PortatoreNon Portatore 1 / 2 1 / 2prior p. 2 / 10 1 condit. p. ½ x 2/10= 2/20 ½ x 1=1/2joint p. 1/101/2 1/10 + 1/21/2 + 1/10post. p. = 1/6 (16.6%) = 5/6 (83.3%) • Test ai membri affetti • conferma della diagnosi • esclusione certa dei non • portatori • definizione del RR della • probanda • SCA : • che tipo di ereditarietà • qual’è la probabilità di essere • portatore per il padre e per la • probanda • è possibile eseguire un test • genetico ? A che scopo ? • a chi offrirlo Autosomica Dominante Probanda: 1/6 x ½ = 1/12(8.3%)
Malattia (9 forme) Locus Ripetizioni normali Ripetizioni patologiche Frequenza tra le ADCA (Italia) Caratteristiche distintive segni piramidali, neuropatia periferica SCA1 6p23 CAG/6-44 39-83 21% movimenti saccadici lenti, neuropatia periferica SCA2 12q24.1 CAG/14-31 36-400 23% nistagmo, retrazione delle palpebre, fascicolazioni amiotrofiche SCA3 14q21 CAG/13-43 55-86 <1% atassia episodica, decorso molto lento SCA6 19p13.1 CAG/ 4-18 (19)20-33 <1% perdita della vista con neuropatia SCA7 3p21.1-p12 CAG / 4-19 37->300 <1% DTR vivaci e diminuzione della sensibilità vibratoria SCA8 13q21 CTG/ 15-50 80-250 <1% attacchi epilettici occasionali SCA10 22q13 ATTCT/ 10-22 800-4500 assente tremori precoci, demenza tardiva SCA12 5q31 CAG / 6-26 66-78 assente SCA17 deterioramento mentale 6q27 CAG/ 30-42 45-63 <1%
SCA : 13 loci noti Malattia Locus N° di famiglie descritte Caratteristiche distintive SCA4 neuropatia assonale sensitiva >5 (japan/scand.) 16q22.1 SCA5 insorgenza precoce, decorso lento 11p12 1 (Lincoln) SCA9 X (riservato) SCA11 lieve, rimane la deambulazione 15q14 -q21 2 (U.K.) SCA13 lieve ritardo mentale, bassa statura 19q13 1 (french) SCA14 Yellow: loci with published markers mioclono assiale precoce 2 (japan/english) 19q13.4 SCA15 3pter-24.2 1 (australian) atassia pura e progressione lenta SCA16 neuropatia sens., atrofia musc. 8q22-24 1 (japan) SCA18 tremore della testa 7q31-32 ? SCA19 1 (dutch) lieve atassia, segni piram., dist. cogn. 1p21-q21 SCA20 riservato SCA21 1 (french) progressione lenta, rigidità, tremore 7p21.3-p15.1 SCA22 1q22 (riservato)
una sola generazione interessata • ramo paterno a materno sani • maschi e femmine affetti • talora segni di consanguineità Ereditarietà autosomica recessiva
Trasmissione ereditaria dei caratteri mendeliani Recessiva : ogni tratto o carattere che si esprima solo nell’ omozigote nel caso di malattia: entrambe le copie del gene mutato devono essere presenti per esprimere un fenotipo affetto Genotipo : eterozigote Fenotipo : portatore sano Genotipo : omozigote Fenotipo : affetto I fenotipi possono essere analizzati e misurati a differenti livelli. Es. malattie metaboliche: gli eterozigoti sono sani ma l’enzima responsabile della malattia può avere nel siero una concentrazione intermedia rispetto a quella dei due stati omozigoti.
a a a Eredità autosomica recessiva Se un genitore è normale (AA) e l’altro è portatore (Aa), allora il 50% in media dei figli sarà omozigote normale (AA), e l’altro 50% sarà portatore eterozigote non affetto (Aa). Non vi sono figli affetti. Nel caso di malattie autosomiche recessive, le lettere minuscole stanno ad indicare i geni che in omozigosi danno la malattia, perché entrambi gli alleli devono essere mutati perché si osservi il fenotipo patologico. Qual’è la probabilità di avere un figlio affetto se entrambi i genitori sono portatori?
Eredità autosomica recessiva Se entrambi i genitori sono portatori sani (Aa), in media il 25% dei figli sarà omozigote normale (AA), il 50% sarà eterozigote non affetto (Aa), e il 25% sarà omozigote affetto (aa). Se un genitore è un portatore sano (Aa) e l’altro è affetto (aa), allora il 50% in media dei figli sarà eterozigote non affetto (Aa), e l’altro 50% sarà omozigote affetto (aa).
Eredità autosomica recessiva • gli individui affetti sono omozigoti(o eterozigoti composti) • nella maggior parte dei casi, entrambi i genitori sono portatori sani – in media, 1 su 4 figli è affetto • la trasmissione non dipende dal sesso • matrimoni tra individui affetti e non affetti genera solo figli eterozigoti sani – a meno che il partner non affetto sia eterozigote • più è rara la malattia, più è probabile che l’individuo affetto sia figlio di genitori tra loro consanguinei (inbreeding)
Aa Aa ? aa AA+Aa Qual è la probabilità di avere un figlio affetto per III-2 ? Probabilità di essere portatore: ½ Probabilità che la coniuge sia portatrice: f portatori nella popolazione ½ x f x ¼ III-2
Conoscendo la frequenza degli affetti : q2 (aa) p = 1 - q2 2pq (f portatori) = 2 (1 - q2 ) q2 Frequenza (f) dei portatori nellapopolazione generale Legge di Hardy-Weinberg p frequenza allele “normale” (A) q frequenza allele “mutato” (a) i genotipi nella popolazione saranno : p2 + q2 + 2pq = (p + q)2dovep + q = 1
Legge di Hardy-Weinbergp2 : q2 : 2pq La distribuzione binomiale presume che : • la popolazione sia ampia • gli incroci siano casuali • le frequenze alleliche siano costanti - frequenza di nuove mutazioni - fitness riproduttiva degli omozigoti affetti - “deriva genetica” - vantaggi degli eterozigoti - migrazioni
Ay Ax xy Qual è la probabilità di avere un figlio affetto per III-2 ? ½ x f x ¼ AA+Ay Se l’individuo IV-2 ha un genotipo noto: mutX/mutY ? -> testing genetico risolutivo Se III-2 è Ay = RR= 1 x f x ¼ Se III-2 è AA = RR pressochè nullo
A? Ax x? Qual è la probabilità di avere un figlio affetto per III-2 ? ½ x f x ¼ Se nell’individuo IV-2 il test genetico ha permesso di identificare una sola mutazione (mutX) ? Stato genetico di III-2 indeterminato RR= ½ x f x ¼ Se III-1 (coniuge) ha un test genetico negativo : RR = ½ x f (sensibilità del test) x ¼
effetto biologico delle mutazioni in relazione alle caratteristiche funzionali della proteina (prodotto genico) Dominanza o Recessività Dipende da : • caratteristiche della malattia ? • caratteristiche del gene ? • effetto biologico delle mutazioni ?
Malattie ereditarie recessive Mutazioni che inducono una perdita di funzione Gli eterozigoti (portatori) sono normali, una riduzione del prodotto proteico del 50% viene tollerato se il rimanente 50% è sufficiente per una funzione normale Esempio: tratto falcemico, bAbS Gli omozigoti sono affetti perché non viene prodotta proteina o quella che è prodotta non funziona normalmente Esempio: anemia falciforme, bSbS difetti enzimatici recessivi, fibrosi cistica
Mutazioni delle “serpine”(inibitori delle serin-proteasi) a1-antitripsina anti-elastasi dei neutrofili antitrombina anti-proteasi della coagulazione inibitore di C1 anti-attivazione del complemento inibitori della plasmina anti-fibrinolisi
Mutazioni dell’ a1-antitripsina (AR)(inibitore dell’elastasi dei neutrofili) mutazioni “loss of function” : ins o del (frame shift) deficit di enzima circolante eccesso di attività dell’elastasi leucocitaria enfisema mutazioni missensodel”sito reattivo” : Met358Arg conversione in antitrombina malattia emorragica fatale mutazioni missensonella”regione mobile o b sheet” : Glu342Lys polimerizzazione della serpina accumulo negli epatociti (ER) fibrosi portale cirrosi ed epatocarcinoma (colangiocarcinoma)
Connessina 26GJB2 (Gap Junction Beta 2) • proteina di 226 aa • esamero (connessone): canale del potassio • espressione: stria vascolare, legamento spirale, tra le cellule di sostegno nella coclea, cute • elevato contenuto di K+ nell’endolinfa
Connessina 26GJB2 (Gap Junction Beta 2) 35DG In Italia : in omozigosi o in eterozigosi composta nel 40% circa di tutti i casi di sordità congenita non sindromica recessiva frameshift e stop
Connessina 26GJB2 (Gap Junction Beta 2) C202F W44C Alleli dominanti : sordità post-linguale progressiva AD
Connessina 26GJB2 (Gap Junction Beta 2) G59A D66H R75W • Alleli dominanti : • Sindrome di Vohwinkel : • sordità congenita AD • ipercheratosi palmo-plantare
Fibrosi cistica (AR, 1/2500) • malattia cronica polmonare • ostruzione diffusa delle vie aeree di piccolo calibro • infezioni polmonari ricorrenti, fibrosi, cisti, ascessi • anomalie gastrointestinali e malassorbimento • Ileo da meconio (15-20%) • insufficienza pancreatica, pancreatite cronica (92%) • malattia epato-biliare cronica (6%) • ostruzione intestinale, prolasso rettale • malassorbimento (vit. liposolubili), ritardo di crescita • azoospermia ostruttiva • assenza dei vasi deferenti • azoospermia, ridotto liquido seminale, pH<7 • sindromi da perdita di Sali • sopravvivenza: 31.6 aa (56aa ps – 20-25aa pi)
Fibrosi cistica (AR, 1/2500) • Tripsinogeno immunoreattivo • screening neonatale • Test del sudore • Cl > 60mEq/L in due separati dosaggi • in una quantità di almeno 75mg (30 minuti) • Potenziale nasale di trans-membrana • dopo i sei anni (potenziale più negativo) • Analisi genetica per la ricerca di 25-31-33 mutazioni “frequenti” • Ricerca di mutazioni di secondo livello
Fibrosi cistica (AR, 1/2500) 7q31.2 gene CFTR 27 esoni mRNA: 6128 b 1480 aa
Mutazioni CFTR e Fibrosi cistica 230 kb 27 esoni 1480 aa proteina ABC ATP-binding cassette
1/112 1/112 + 27/28 Test OLA (31-33 mutazioni): sensibilità del 75% f = 1/28 RA = 1/28 • 1/28 • 1/4 = 1/3100 ~ Test OLA negativo 3/4 P mut+ (75%) 1/4 P mut- (25%) =1/109 RA = 1/109 • 1/109 • 1/4 = 1/47500 ~
XXXXXXXXXXXXXXXXXXX TTTCTTTTATAGTAGAA ATTATGCCTGGCACCATTAAAGAAAATATCATCTTTGGTGTTTCCTATGATGAATATAGA XXXXXXXXXXXXXXX TAATTTCTTTTATAGTA ATTATGCCTGGCACCATTAAAGAAAATATCATTGGTGTTTCCTATGATGAATATAGA DF508 e test OLAOligonucleotide Ligation Assay CFTR cDNA Esone 10 1561 CCTTCAGAGGGTAAAATTAAGCACAGTGGAAGAATTTCATTCTGTTCTCAGTTTTCCTGG 477 -P--S--E--G--K--I--K--H--S--G--R--I--S--F--C--S--Q--F--S--W- 1621 ATTATGCCTGGCACCATTAAAGAAAATATCATCTTTGGTGTTTCCTATGATGAATATAGA 497 -I--M--P--G--T--I--K--E--N--I--I--F--G--V--S--Y--D--E--Y--R- 1681 TACAGAAGCGTCATCAAAGCATGCCAACTAGAAGAGGACATCTCCAAGTTTGCAGAGAAA 517 -Y--R--S--V--I--K--A--C--Q--L--E--E--D--I--S--K--F--A--E--K- 1741 GACAATATAGTTCTTGGAGAAGGTGGAATCACACTGAGTGGAGGTCAACGAGCAAGAATT 537 -D--N--I--V--L--G--E--G--G--I--T--L--S--G--G--Q--R--A--R--I- Esone 11 Seq. wt Seq. mut
XXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXX TTTCTTTTATAGTAGAAACCACAAAGGATACTACTT ATTATGCCTGGCACCATTAAAGAAAATATCATCTTTGGTGTTTCCTATGATGAATATAGA XXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXX TAATTTCTTTTATAGTAACCACAAAGGATACTACTT ATTATGCCTGGCACCATTAAAGAAAATATCATTGGTGTTTCCTATGATGAATATAGA Reazione di Ligasi e amplificazione mediante PCR con primers specifici per le code: Pannello di prodotti di PCR di dimensioni diverse a seconda della specificità dell’oligonucleotide separati con elettroforesi capillare Es. F508 wt : 74 bp DF508 : 70 bp
1958 1965 1986: diagnosi di azoospermia associata ad assenza dei vasi deferenti. Prelievo di spermatozoi e tentativi di fecondazione assistita falliti….in attesa di ICSI. Marito:CFTR D1270N-5T introne 8 Moglie:CFTR negativo per le 31 mutazioni più frequenti e analisi di 10 esoni in DGGE rischio residuo di essere portatrice: 1/200 Rischio per la coppia di avere un figlio affetto da una forma lieve di fibrosi cistica o CBAVD di 1/400
Mutazioni CFTR e CBAVD • Sono spesso presenti (70%) segni sub-clinici di fibrosi cistica: • test al sudore alterato (> 40 mEqu/l) • ostruzione nasale, polipi nasali, sinusiti • infezioni respiratorie ricorrenti
Mutazioni dominanti • Mutazioni dominanti negative • proteina che non funziona • e che inibisce o interferisce con la funzione della proteina normale codificata dall’allele sano • tipico delle proteine multimeriche
Osteogenesi imperfetta (AD) • Collagene tipo I • 2 molecole di Col a 1 (17q) • 1 molecola di Col a 2 (7q) • (338 ripetizioni GXY)
Osteogenesi imperfetta (AD) OI lieve (tipo I) statura normale, scarse deformità sclere blu e sordità, fratture OI letale-grave (tipo II-III) fratture multiple accorciamento delle ossa lunghe deformità ossee, sordità mut de novo in Gly 5-10% : mosaicismo germinale OI moderata (tipo IV) bassa statura, deformità osteoporosi, dentinogenesi imperfetta