Download
1 / 43
430 likes | 556 Views
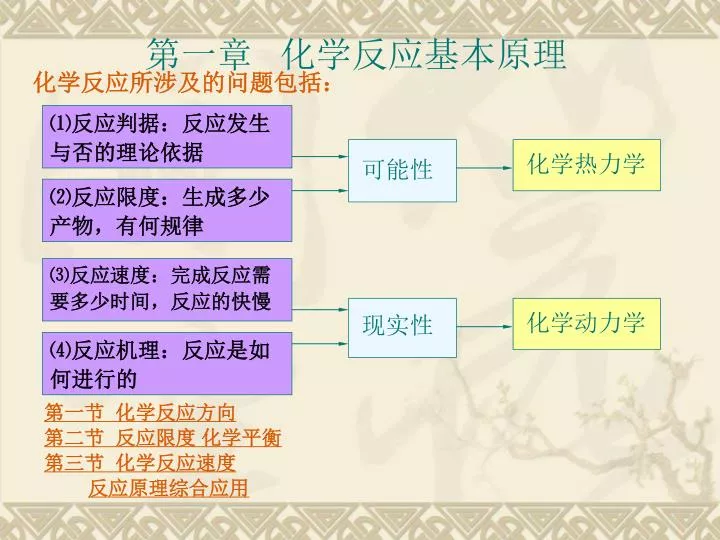
⑴反应判据:反应发生与否的理论依据. 化学热力学. 可能性. ⑵反应限度:生成多少产物,有何规律. ⑶反应速度:完成反应需要多少时间,反应的快慢. 化学动力学. 现实性. ⑷反应机理:反应是如何进行的. 第一章 化学反应基本原理. 化学反应所涉及的问题包括:. 第一节 化学反应方向 第二节 反应限度 化学平衡 第三节 化学反应速度 反应原理综合应用. 热力学简介 它是研究能量转换过程中所遵循规律的一门学科。于19世纪中叶,蒸汽机的发明和应用,研究热和机械功之间的转换关系形成,后来研究范围推广到各种能量之间的相互转换中。.

E N D
⑴反应判据:反应发生与否的理论依据 化学热力学 可能性 ⑵反应限度:生成多少产物,有何规律 ⑶反应速度:完成反应需要多少时间,反应的快慢 化学动力学 现实性 ⑷反应机理:反应是如何进行的 第一章 化学反应基本原理 化学反应所涉及的问题包括: 第一节 化学反应方向 第二节 反应限度 化学平衡 第三节 化学反应速度 反应原理综合应用
热力学简介 它是研究能量转换过程中所遵循规律的一门学科。于19世纪中叶,蒸汽机的发明和应用,研究热和机械功之间的转换关系形成,后来研究范围推广到各种能量之间的相互转换中。 特点:1、基础是两个经验定律; 2、结论具有统计意义:是讨论大量质点的平均行为,不 涉及物质的微观结构和过程机理。 局限性:不能讨论过程如何进行和进行的快慢; 结论不能说明过程进行的机理和快慢。
热力学一些专门术语 体系(系统):研究对象。 环境:与体系紧密联系的部分。 体系分类: (1) 敞开系统:系统与环境之间有物质和能量的交换; (2) 封闭系统:系统与环境之间有能量交换但无物质交换; (3) 隔离系统:也称孤立系统,系统与环境之间既无物质交换 也无能量交换。 P V n T均为状态函数 状态:体系不再随时间变化的情形(平衡态) 状态函数:描述体系状态性质的函数(物理量) 状态函数特征: 状态一定值一定,殊途同归变化等,周而复始变化零。
热力学一些专门术语 热量:发生变化时,体系与环境之间因温差传递的能量形式。它与变化相关联。规定:体系从环境吸热为正。 功:除热以外的其他能量传递形式。规定环境对体系做功为正 思考: Q和W是不是状态函数? 三种过程:恒温过程、恒容过程和恒压过程。 恒温过程 恒容过程 恒压过程 内能:体系内部能量总和——热力学能。 热力学第一定律——能量守恒 即: Q= △U - W 热力学第二定律: 不能从单一热源吸热使之完全变为功而不发生其他变化
第一节 化学反应方向 可逆反应在某条件下,反应方向如何判断? 能量最低原理与自然现象 1-1自发过程 一定条件下,无需外力作用能自动进行的过程。 “自发”不含“快速”之意 “非自发”不等于“不可能” 自由能G 最小原理 能量最 低原理 热效应 焓变 熵增 原理 计算 处理 计算
1-2化学反应热效应,反应焓变 • 1.反应热 • 一定温度下,只对抗外压作膨胀功时,反应吸收或放出的热量。 • 恒容反应热 QV=△U(由第一定律得到) • 恒压反应热 QP=△H • 2.焓(H)——新物理量 • 物质的又一种能量形式。 H≡ U + PV • 如何理解 QP=△H
恒温恒压下,反应物、产物各自有不同的焓。当反应物的焓比产物的焓高时,由反应物转变为产物,就要释放出那多余的部分(△H),以热量的形式释放。恒温恒压下,反应物、产物各自有不同的焓。当反应物的焓比产物的焓高时,由反应物转变为产物,就要释放出那多余的部分(△H),以热量的形式释放。 △H = H产物 - H反应物 △H>0 QP>0 吸热 △H<0 QP<0 放热 H N2+3H2 H △H 2NH3
3.Hess定律 反应的热效应只与始终态有关,与变化途径无关 反应一步完成的热效应等于分步进行的各步热效应之和 注意: • 条件相同的反应或聚集状态相同的同种物质才能相消或合并。 • 反应式乘(或除)以某数,相应的热效应也要同乘(除)以该数。 应用——作业
4、热化学方程式(△H⊙表示反应热) 思考: ①对同一反应,反应式书写不同,△H⊙值有无变化——作业 ②△H⊙的单位为KJ/mol,mol-1表示什么? ③⊙代表什么?
⊙代表标准态——物质状态的参比基准线 规定:标准压强下物质的确切聚集状态。 [注] 对温度没有规定。 [比较] 气体标准状态。 说明: ① 气体的标准态:P⊙ = 1.013×105 ② 溶液中溶质的标准态:标准压强下,溶质的浓度为 1mol/L, 即 C⊙ =1mol/L ③ 液体和固体的标准态: P⊙下的纯物质。
5、反应热的计算 ①、原则——Hess定律 [问题] 能否通过较少的实验数据获得任意化学反应的热效应? [分析] 反应 A + B C + D △H⊙ = H ⊙产物 - H ⊙反应物 绝对法难确定△H⊙ ,可用相对法解决
②、标准生成焓——△fHm⊙ 定义:温度T、标态时,由指定单质生成1mol某物质(以化学式表示)的焓变。 [说明] 指定单质:多为元素的最稳定单质,状态亦指定。 生成反应:指定单质反应生成1mol生成物的过程 [推论]: 指定单质△fHm⊙=0 △fHm⊙非绝对值 熟悉元素的指定单质、物质的生成过程。 H, Na, K, C, O, N, F, Cl, Br, I 写出生成过程:CO2 ,MgO, KBr, NaI
反应物 产物 ②① 各种指定单质 [问题] 已知物质的△fHm⊙ ,如何计算反应的△H⊙? [设计] [思路] 过程Ⅰ 由各指定单质直接生成产物; 过程Ⅱ 由各指定单质先生成反应物,然后 反应物再转化为产物 必有:过程Ⅰ热效应 = 过程Ⅱ热效应 [公式]△H⊙ =∑△fHm⊙ 产物 -∑△fHm⊙ 反应物
1-3标准态时反应的方向 许多放热反应为自发过程: 如:3Fe(s) + 2O2(g) → Fe3O4(s)△H⊙ =-1120KJ 2H2 + O2 →2H2O(g)△H⊙ =-483.6KJ 但有些吸热过程也能自发进行: 如:常温常压下H2CO3(aq) → H2O() + CO2(g) 该反应△H⊙= +19.3KJ,但自发进行。 [为什么] 仔细考察这类过程有共同的特点:混乱程度增大。 所以,体系的混乱度也对过程的方向有影响: 有序到无序利于自发,反之不利于自发。
1、熵——S 表征体系组成微粒分布混乱程度的函数。 S值越大,混乱度越大,有序性越差。 规律:熵值 Ⅰ、固体<液体<气体 Ⅱ、混合物>纯净物 Ⅲ、S随温度升高增大 热力学第三定律: 各种纯化学物质的最完整晶体,在0K时熵值为零粒子的热运动完全停止,内部排列达到最有序程度。即S0K=0 设: A0K → ATK △S = ST -S0 = ST ST为绝对值,称绝对熵。
2、标准熵 定义:1mol纯物质在标准态时的熵值。ST⊙ 单位:J·mol-1·K-1 [比较] △fH⊙ 与 ST⊙ ①ST⊙为绝对值, △fH⊙为相对值; ②指定单质△fH⊙ =0,而ST⊙≠0。 3、 △S⊙计算 与△H⊙计算原则、方法一样。 △S⊙ =∑ S⊙产物 -∑ S⊙反应物 [分析]:前述自发吸热过程都是△S⊙>0 故 自发过程的方向受两大因素制约:△S⊙、△H⊙
小结 ① △H⊙ <0 , △S⊙ >0 自发; ② △H⊙ >0 。 △S⊙<0, 非自发。 ③ △H⊙ <0 , △S⊙ <0 ④△H⊙ >0 。 △S⊙ >0, [美]Gibbs提出:恒温恒压下,△H和△S可综合成一个新的函数变量,记为△G称Gibbs自由能变量。 关系: △G =△H-T△S * △G⊙ =△H⊙ -T△S⊙ * 4、自由能G(又一新能量形式) 意义不讨论。
① 最小自由能原理: 恒温恒压下 △G<0 自发; △G>0 非自发 △G⊙ <0 标态自发; △G⊙>0 标态非自发 由关系△G⊙ =△H⊙ -T△S⊙ 假设△H⊙ 、△S⊙不随温度变化,可估计自发反应进行的温度范围。 ②△G⊙ 计算(与△H⊙相似) Ⅰ、由 △fG⊙——标准生成自由能计算 Ⅱ、由△G⊙ =△H⊙ -T△S⊙ 计算 本节小结: 介绍了化学热力学的初步知识,主要讨论了焓、熵、自由能等状态函数,其意义不深究,解决了反应方向的判据。重点在于应用: Ⅰ、 △G⊙、△H⊙、△S⊙计算 Ⅱ、用△G⊙判断自发反应方向。
例1:据美国《化学及工程新闻》报道,在常温常压下,利用某种催化剂由水与氮气反应制取氨。设想可能是下列反应吗?例1:据美国《化学及工程新闻》报道,在常温常压下,利用某种催化剂由水与氮气反应制取氨。设想可能是下列反应吗? 2N2(g)+6H2O(l)==4NH3(g)+3O2(g) △fHm⊙ 0 -286 -46 0 ST⊙ 191 70 192 205 例2:能否用下列反应合成酒精? 4CO2(g)+ 6H2O(l)== 2C2H5OH(l)+7O2(g) △fHm⊙ -393 -286 -278 0 ST⊙ 214 70 161 205
第二节 反应限度 化学平衡 反应方向确定后,反应进行到什么程度才停止呢? 2-1 反应限度的判据 1、能量判据 [分析] 恒温恒压下,根据最小自由能原理,反应之所以自发进行,是因为△G<0 。 随着反应进行,必有∑G产增大,∑G反降低。故有△G →0趋势。 当△G=0时,反应失去“推动力”,宏观上反应“不再进行”,即自发反应进行到△G=0而“停止”。 反应限度的能量判据: △G=0
[比较] 高中知识对可逆反应,最终有∨正=∨逆,正反应单位时间内消耗的反应物必然为逆反应所补偿,宏观上各组分的量不再随时间而变,达到化学平衡。 • ∨正=∨逆为反应限度的质量判据。 2、化学平衡——反应的限度 恒温恒压下,反应达到∨正=∨逆或△G=0时的状态 • 化学平衡建立的条件: 恒温恒压, ∨正=∨逆△G=0 • 平衡建立的标志: 各组分的数量不再随时间而变。
化学平衡的特征(定性): ①动态平衡。 ②是恒温恒压封闭体系中可逆反应进行的最大限度或最终状态。 [问题]化学平衡的定量特征,平衡组成之间的关系? 3、化学平衡定律 ①活度商(反应商)Qi 反应 aA + bB cC + dD
② Van’t Hoff恒温方程式: △G=△Go+RTlnQi 平衡时, △G=0 △Go= -RTln(Qi)平 [分析] 该式将反应的标准自由能变与平衡时各物质的组成关系联系起来了。 式中R,T,△Go均为常数,(Qi)平必为常数。写为Ko ③平衡常数——质量作用定律 一定温度下,反应达到平衡,产物浓度以反应方程式计量系数为乘幂的乘积与反应物浓度以计量系数为乘幂的乘积之比为常数。 一定温度下,每个平衡反应都有它自己特征的平衡常数,这是化学平衡的定量特征。
④ Ko的意义、性质: • 意义:反应了平衡混合物中产物所占的相对比例,它很好的反映出反应进行的程度。 表明了一定温度下反应平衡的条件: Q = Ko平衡 Q > Ko反应自右向左进行 Q < Ko反应自左向右进行 • Ko与 Q异同: 都对应相同的反应式,但Q为任意态下的值,ko为平衡时的值。 • 性质:它仅为温度的函数,与浓度无关,不随平衡组成而变,与达到平衡的途径无关。 ⑤平衡常数的表示法及书写规则(自学为主) 思考:“合成氨反应在500℃时的平衡常数为0.0001”说法。
2-2多重平衡规则——K的组合 一定温度下,若反应可表示为多个分反应之和或差,则该反应平衡常数等于各分反应平衡常数之积或商。 即平衡常数与达到平衡的途径无关。 [分析]∵△Go= -RTlnKo 若:总反应=反应1+反应2 △Go总=△Go1+△Go2 -RTlnKo=-RTlnKo1-RTlnKo2 ∴KO=KO1×KO2 [注] 各反应必须是同一温度,各物质集聚状态相同才能组合。
2-3 应用——平衡计算 [内容] ko的确定及平衡组成的有关计算 1、K的确定 Ⅰ.根据△Go= -RTlnKo Ⅱ.多重平衡规则 Ⅲ.实验测定 2、计算平衡组成 平衡转化率:简称转化率(离解率、分解率) 平衡时已转化的某反应物的量与转化前该物质的量之比。 它也反映了反应进行的程度
例:已知反应CO2(g)+H2(g)==CO(g)+H2O(g) T=1473K时Kc=2.3,求: ①当CO2、H2起始浓度均为0.01mol/L ②当CO2起始浓度为0.01mol/L,H2起始浓度为0.02mol/L 两种情况下。CO2的转化率。 [小结]: K和转化率都可表示反应进行的程度,但转化率与反应物起始浓度和反应温度有关,K而仅为温度的函数 注意解题步骤。
2-4 平衡移动 动态平衡:改变平衡条件,使△G≠0,平衡破坏。 平衡移动:反应从旧平衡点转变到新平衡点的过程 • 浓度、压强、温度等因素都可使平衡发生移动。 [分析]: △G=△Go+RTlnQi 且△Go=-RTlnK =-RTlnk+RTlnQi =RTln(Qi/K) [讨论]①恒温下平衡时 Qi=K△G =0 Qi<K△G<0 ,正向移动至Qi*=K Qi>K △G>0,逆向移动至Qi#=K
②仅改变温度,旧平衡 T1 Qi=K1 改变温度至T2 Qi≠K2 平衡被破坏 移动至 Qi*=K2 关键是K如何改变 1、浓度对平衡的影响 [定量计算] 例 CO+H2O==CO2+H2T=1073 K,密闭容器中2molCO和H2O混合,平衡时CO转化率为50%。若再加入3mol H2O,问平衡能否维持? 新平衡时CO的转化率为多少? [实际应用] 利用廉价原料或不断移走产物,以提高某反应物的转化率。
2、压强的影响 [定性]改变压强,可导致体积的变化,而影响化学平衡。较复杂。 ①对无气体参与的固液反应体系,几乎无影响 ②对有气体参加的反应压力的变化,可能有影响。 [分析]反应 aA(g) + bB(g) cC(g) + dD(g) 平衡时 ∵PV = nRT 仅改变体积 P∝V-1 若改变体积为原来的m倍,则P’=P×m-1
改变体积为原来的m倍,则总压及各物质均为旧平衡的m-1倍。改变体积为原来的m倍,则总压及各物质均为旧平衡的m-1倍。 则: • Qp=Kp×(1/m)(c+d-a-b) 令△n=c+d-a-b • = Kp ×(1/m)△n • [讨论] • 若△n=0 反应前后气体分子数无变化,改变体积Qp=Kp不影响平衡。 • 若△n≠0 • 当△n>0 0<m<1即总压增大,Qp>Kp 逆向移动 • m>1 即总压减小, Qp<Kp 正向移动 • 当△n<0 0<m<1即总压增大,Qp<Kp 正向移动 • m>1 即总压减小,Qp>Kp 逆向移动
例:已知反应PCl5(g)==PCl3(g)+Cl2(g)在523K时,将0.7mol PCl5注入2.0L的密闭容器中,平衡时有0.5mol PCl5分解了。在达平衡后,将密闭容器的体积减小为1L,问平衡能否维持,各组分的压强和PCl5的分解率为多少。
3、温度的影响 K是温度的函数,改变温度,K变,平衡被破坏。 [分析] ∵△Go=-RTlnK △Go =△Ho -T△So 若忽略温度对△Ho 和△So的影响 处理:lnK= △So/R-△Ho/RT 讨论: 4、催化剂对化学平衡的影响 [结论] 催化剂只改变反应速度,对已达到平衡的反应,加入催化剂,同等程度改变正逆反应速度,故对平衡无影响。 即催化剂不能改变平衡状态。 [总结] Le.Chatelier原理 改变影响平衡的某一因素,平衡就沿着减弱这种影响的方向移动,直至新平衡。
第三节 化学反应速度 反应速度与热力学之间没有直接的联系。为什么? 说明:计算常温反应2H2 + O2 2H2O(l)的平衡常数。 思考:该条件实际反应情况与反应趋势。 3-1 反应速度的表示(自学为主) 1、定义:一定条件下,反应物转变为产物的快慢。 2、表示:单位时间某物质的浓度变化。 注意:①可用不同物质的浓度变化表示反应速度; ②其数值不一定相等,但意义相同,有关系 ③单位:mol/L·s 对大多数反应,物质浓度随时间的变化不是呈 线性关系故有平均速度和瞬时速度之分
3、速度测定 ①平均速度 ②瞬时速度:实验测定物质浓度随时间变化的数据,处理曲线某点切线的斜率。 [问题]为什么反应速度千差万别? 3-2 速度理论 1、碰撞理论(自学为主) 要点:①反应进行的先决条件:分子相互碰撞; ②有效碰撞:发生反应的碰撞; ③活化分子:发生有效碰撞的分子,它比一般分子的能量高。一定条件下,分子能量分布一定。 ④活化能:活化分子具有的平均能量与反应物分子的平均能量之差。(Ea) ***
活化分子平均能量 Ea A+B △H C+D 反应进程 活化能越大,反应逾越的能量障碍越大,反应速度越小。 如反应N2+3H2 2NH3△Ho=-92.38KJ/mol △Go =-33.28KJ/mol 常温常压自发,但此条件下,正反应的 Ea=326KJ/mol 所要逾越的“能量障碍”大。活化分子数少,速度小。 2、过渡状态理论简介 要点:①反应物分子碰撞后经过一个中间过渡状态,形成活性集团(称活化络合物),再分解为产物(或反应物)。
②活化络合物与反应物(产物)存在能垒,称正反应(逆反应)的活化能。②活化络合物与反应物(产物)存在能垒,称正反应(逆反应)的活化能。 活化能的本质,学术界至今尚未取得统一的认识。 3-3影响反应速度的外部因素 浓度、压强、温度、催化剂 一、浓度(压强)的影响 定量关系 1、速率方程 反应物浓度与反应速度的数学关系式。 反应 aA(g) + bB(g) cC(g) + dD(g) V∝CAαCBβ= k CAαCBβ [说明] k 为比例常数,称速率常数 α、β为反应分级数, α+β为反应总级数
2、反应级数:反映了浓度(压力)对速度的影响程度。2、反应级数:反映了浓度(压力)对速度的影响程度。 3、速率常数的意义和性质 [分析] 当反应物浓度均为1mol/L时V=k ①一定温度下, k值取决于反应本性,不同反应k不同,k反映V的内因。 ②k 值不随浓度、压力而变,与温度、催化剂有关。 ③k 单位与反应级数有关。 [强调] 反应级数与反应计量系数并非一回事,不一定相符合。反应级数是实验值,可为整数、分数、小数。 对一般反应,级数不能直接从计量系数导出。 要深入了解浓度对反应速度的影响,必须涉及反应经历的具体历程。(本课程不讨论)
二、温度的影响 升高温度,加速反应,早就被人所知。 [定性分析] 浓度不变,分子总数不变,温度升高,部分分子获得能量,变成活化分子,即活化分子百分数增加,有效碰撞增加,反应速度加快。 [定量] Arrhenuis公式: k=Aе-(Ea/RT)或nk= nA-(Ea/RT) A ——常数,称频率因子或指前因子 Ea——实验活化能 R——气体常数 表明: k与T呈指数变化关系,T微小的变化, k就有较大变化。应用:计算,注意单位换算。 [思考] 对不同反应, k随温度的变化规律。
活化分子平均能量 Ea A+B +K △H AK+B C+K 反应进程 三、催化剂的影响 [概念] 改变反应速度,自身在反应前后的质量和化学组成不变的组分。 正、负催化剂;酶、触媒、防老剂等。 催化剂对V的有效影响是浓度、温度无法比拟的。 [研究表明] 催化剂通过改变反应途径,改变反应活化能实现对反应速度的影响,它参与了反应。 [催化剂特性] ⑴ 选择性; ⑵ 不能起动反应,不能使非自发反应发生 ⑶ 不能改变反应的平衡常数
本章总结 解决了化学反应所涉及的问题: ⑴反应判据 ⑵反应限度 ⑶反应速度 ⑷反应机理线索(思路) 反应原理综合应用(选讲) [实例分析] 水煤气变换条件分析 CO(g)+H2O(g)→ CO2(g)+H2(g) 反应条件: 投料比:CO : H2O=1 : 5~8; 总压: 5~7×105Pa; 温度: 多段处理(高温变换,低温出料)
[方法] 1、方向判断及反应自发进行的温度范围确定; 2、改变转化率的条件确定; 3、改变反应速度的条件确定; 4、其他考虑的因素。 CO(g)+H2O(g)→ CO2(g)+H2(g) ⑴ 反应有关的热力学数据: △H⊙=-41KJ△S⊙ =-42.4 J·K-1 △G⊙ =-28.5KJ 常温常压反应自发。 ⑵ 自发反应温度范围 T< △H⊙/△S⊙ ≈700℃ ⑶ 从平衡移动角度分析 K随温度升高而减小,即CO平衡转化率降低(△H⊙<0); 增加水汽量可提高CO转化率; △n=0,改变压力对平衡无影响。
⑷从反应速度角度考虑 温度升高,速度增大,常温该反应速度小; 总压增加,速度增大,可加速达到平衡; 催化剂使用,提高反应速度; 增加水汽量可提高反应速度。综合反应条件: 解决冲突,考虑经济核算及操作控制。 投料比:CO : H2O=1 : 5~8,过多水汽温度难控制。 总压:5~7×105Pa 温度:多段变换处理。 高温变换,低温出料: 高温提高速度,迅速建立平衡,然后降温平衡移动提高转化率。
