Download

1 / 26
260 likes | 503 Views
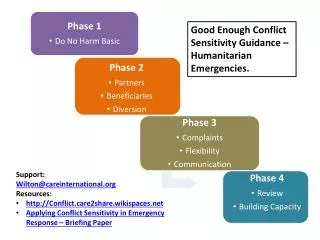
Draft Guidance for Industry: CGMPs for Phase 1 INDs. Joseph C. Famulare, Director Division of Manufacturing & Product Quality Office of Compliance, CDER, FDA Advisory Committee for Pharmaceutical Science Manufacturing Subcommittee 20-21 July 2004. Background.

E N D
Draft Guidance for Industry: CGMPs for Phase 1 INDs Joseph C. Famulare, Director Division of Manufacturing & Product Quality Office of Compliance, CDER, FDA Advisory Committee for Pharmaceutical Science Manufacturing Subcommittee 20-21 July 2004
Background • 501(a)(2)(B) requires all drug products be manufactured in accordance with current good manufacturing practice (CGMP) • CGMP Regulations, published in 1978 and codified in 21 CFR 210 and 211 primarily directed to commercial manufacturing of approved drugs and biologics
Background (cont’d.) • Preamble to 1978 CGMP regulations: • states that CGMP regulations are applicable to preparation of any drug product for administration to humans or animals • indicated FDA’s intent to publish additional regulations specific to investigational clinical studies
Background (cont’d.) • The 1991 Guideline for preparation of investigational new drug products: • does not adequately cover all manufacturing situations • does not fully address Agency expectation that an incremental approach to CGMP compliance is acceptable for investigational products
Background (cont’d.) • Draft guidance for Phase 1 INDs and complementing regulation: • articulates FDA’s intent to implement an incremental approach to CGMP compliance for clinical investigational products • recognizes that some controls and the extent of controls differ between investigational and commercial manufacturing, as well as phases of clinical studies
Guidance – Development Principles • Developed by Agency workgroup (CDER, CBER, ORA) composed of compliance staff, CMC reviewers, and inspectors • Utilize risk-based approach • available knowledge • emphasis on safety • General CGMP
Scope • Draft Guidance • Applies to investigational new drug and biological drug products used during phase 1 clinical studies • Specifically addresses phase 1 studies designed to assess tolerability or feasibility for further drug development work • Some phase 1 studies are excluded (e.g., drug metabolism, structure-activity relationships, food interaction)
Scope (cont’d.) • Draft Guidance: • provides direction for special production situations (i.e. microdose, multi-product, multi-lot) and specific product types • intended to serve as a companion to other guidance describing chemistry, manufacturing and control info submitted in Phase 1 INDs, and CGMPs for the manufacture of APIs (ICH-Q7A)
Recommendations for complying with CGMP Requirements • Utilize appropriate quality control (QC) standards, i.e.: • well defined procedures • adequately controlled equipment • accurate recording of all data • Application of QC standards leads to implementation of CGMPs consistent with good scientific methodology
Recommendations for complying with CGMP Requirements (cont’d.) • Use available technology and resources to facilitate product development, CGMP compliance, and lessen CGMP burden, i.e.: • disposable equipment and process aids • prepackaged water for injection (WFI) and sterilized containers • contract manufacturing and testing facilities
Recommendations for complying with CGMP Requirements (cont’d.) • Prevent contamination and cross-contamination: • evaluate production environment to identify potential hazards • ensure that substances (i.e. chemicals, adventitious agents) from previous or concurrent research or production are removed
General CGMP Requirements • Personnel: • education, experience and training, (or any combination of the three) to perform assigned functions, e.g. training to include GMPs outlined in guidance
General CGMP Requirements (cont’d.) • Quality Control Function: • is established for every producer of IND products • responsibilities are documented in writing and include: • examination of components, containers, closures, in-process materials, packaging and labeling materials • review and approval of production and testing procedures, acceptance criteria • review of completed production records for release or rejection of each clinical batch • is the responsibility of all staff involved in production • in operations with limited staff– QC function may be carried out by the same person performing production (with periodic review by another qualified person)
General CGMP Requirements (cont’d.) • Facilities: • adequate work areas for intended tasks • water of appropriate source and quality • adequate air handling to prevent contamination and cross-contamination
General CGMP Requirements (cont’d.) • Equipment: • in proper working condition, maintained calibrated, cleaned and sanitized at appropriate intervals • appropriate for intended function • should not contaminate or be reactive, additive or absorptive with product • identified and documented in production records
General CGMP Requirements (cont’d.) • Control of Components: • written procedures describing handling and control of components • written, specified attributes or acceptance criteria • COA or other documentation for components to ensure conformance with specified attributes • records of receipt, quantity, supplier’s name, lot number, expiration date
General CGMP Requirements (cont’d.) • Production and Documentation: • records including laboratory and production data detailing components, equipment and procedures used • records of changes in processes and procedures • appropriate microbiological control for production of sterile processed products
General CGMP Requirements (cont’d.) • Laboratory Controls: • tests conducted using established written procedures under controlled conditions • scientifically sound analytical procedures • properly calibrated and maintained analytical lab equipment • initiate stability study to support use of product in clinical investigation
General CGMP Requirements (cont’d.) • Container Closure and Labeling: • Package to protect product from alteration, contamination and damage during storage, handling and shipping • Controlled labeling to preclude mix-ups • Distribution: • Describes the transport of the IND product from the point of production to the patient/subject for consumption
General CGMP Requirements (cont’d.) • Recordkeeping • Retain records related to quality and operation of production processes including: • Equipment maintenance and calibration • Production records and related analytical test records • Distribution records • All quality control functions • Component records • Retain records for 2 years after approval of marketing application or until 2 years after shipment and delivery of the product is discontinued and FDA is notified
Special Production Situations • Screening/Microdose INDs • Multi-Product Facilities • Biological and Biotechnological Products • Sterile/Aseptically processed products
Screening/Microdose INDs • The application of CGMPs controls to Screening IND and Microdose studies should be proportional to the scale and scope of the operation. • Special provisions for lab scale production are provided in the guidance with respect to QC, facility, equipment, and lab control.
Multi-product Facilities • An area or room can be used for multiple purposes and products, provided that: • only one product is produced in an area at any given time • appropriate cleaning and change over procedures are in place to ensure there is no carry-over of materials or products or mix-ups
Biological and Biotechnological Products • Additional safeguards • Some production systems may warrant additional safeguards to protect personnel (e.g., pathogenic microorganisms, some products made from spore-forming microorganisms, live viral vaccines and gene therapy vectors) • Equipment qualification and controls in production assure success of unit operations with safety-related functions (e.g., viral clearance, virus/ toxin attenuation, pasteurization)
Biological and Biotechnological Products (cont’d.) • Retained samples (e.g., drug substance, drug product, intermediates) can be subsequently analyzed and compared to provide assurance of product consistency throughout clinical development. • In-process testing and detailed records ensure product consistency for Phase 1 products which are produced in multiple lots
Sterile/Aseptically Processed Products • Investigational products intended to be sterile require specific precautions, for example: • personnel should be properly trained in aseptic techniques • aseptic manipulation should be conducted in a Class 100 environment e.g. laminar flow hood • controls should be in place to assure appropriate air quality of the aseptic environment